 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLife cycle assessment for all organic chemicals
Mar 15, 2026Chemicals are embedded in nearly every aspect of modern society, yet their production poses substantial sustainability concerns. Achieving a sustainable chemical industry requires detailed Life Cycle Assessment (LCA); however, current assessments face many unknowns due to limited, partly inconsistent, and untransparent data coverage since existing Life Cycle Inventory (LCI) databases account for only a tiny fraction of traded chemicals. Here, we introduce the Chemical RetrosYnthesiS for Transparent Assessment of Life-cycles (CRYSTAL) framework, which automatically generates consistent and transparent LCI data for organic chemicals based on their molecular structure using retrosynthesis and machine-learned gate-to-gate inventories. Using the predictive power of CRYSTAL, we create a consistent database for more than 70000 organic chemicals, comprising over 110000 transparent LCI datasets that quantify both feedstock and energy demands, together with associated auxiliary materials, biosphere flows, and waste flows. From this comprehensive database, we identify 50 key environmental hotspots driving high impacts of organic chemical production across multiple environmental categories and pivotal hub chemicals that are most critical for downstream chemical production. In providing this comprehensive data foundation, the CRYSTAL framework offers systematic guidance for targeted engineering and policy interventions. Its transparent, modular nature is designed to shift chemical LCA from a reliance on "unknown unknowns" to a collaboratively improvable mapping of "known unknowns".
SPT-NRTL: A physics-guided machine learning model to predict thermodynamically consistent activity coefficients
Sep 09, 2022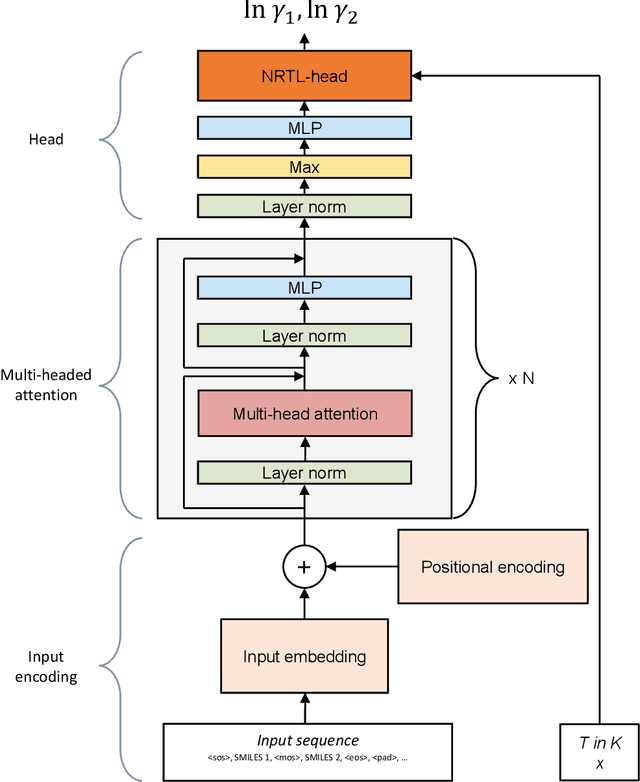
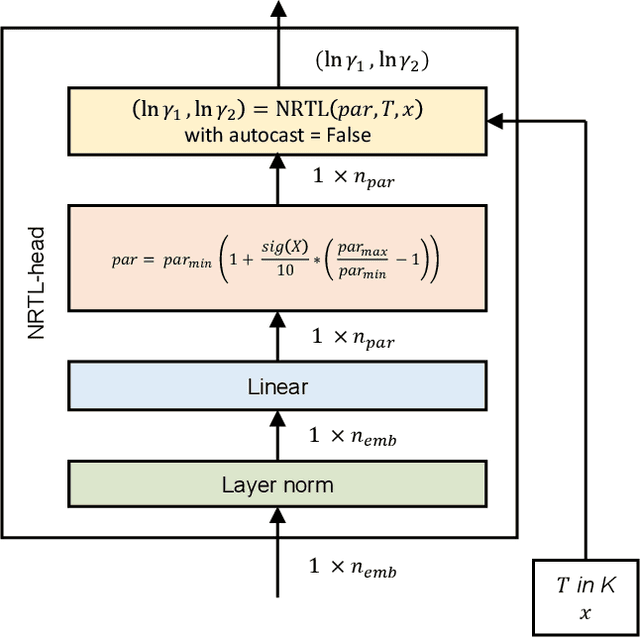


The availability of property data is one of the major bottlenecks in the development of chemical processes, often requiring time-consuming and expensive experiments or limiting the design space to a small number of known molecules. This bottleneck has been the motivation behind the continuing development of predictive property models. For the property prediction of novel molecules, group contribution methods have been groundbreaking. In recent times, machine learning has joined the more established property prediction models. However, even with recent successes, the integration of physical constraints into machine learning models remains challenging. Physical constraints are vital to many thermodynamic properties, such as the Gibbs-Dunham relation, introducing an additional layer of complexity into the prediction. Here, we introduce SPT-NRTL, a machine learning model to predict thermodynamically consistent activity coefficients and provide NRTL parameters for easy use in process simulations. The results show that SPT-NRTL achieves higher accuracy than UNIFAC in the prediction of activity coefficients across all functional groups and is able to predict many vapor-liquid-equilibria with near experimental accuracy, as illustrated for the exemplary mixtures water/ethanol and chloroform/n-hexane. To ease the application of SPT-NRTL, NRTL-parameters of 100 000 000 mixtures are calculated with SPT-NRTL and provided online.
A smile is all you need: Predicting limiting activity coefficients from SMILES with natural language processing
Jun 15, 2022
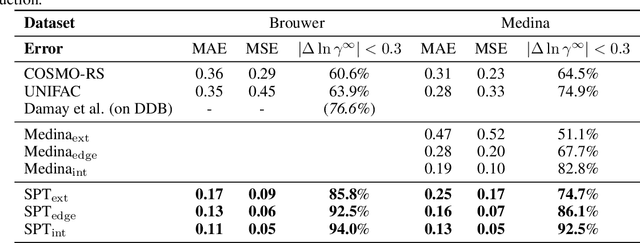
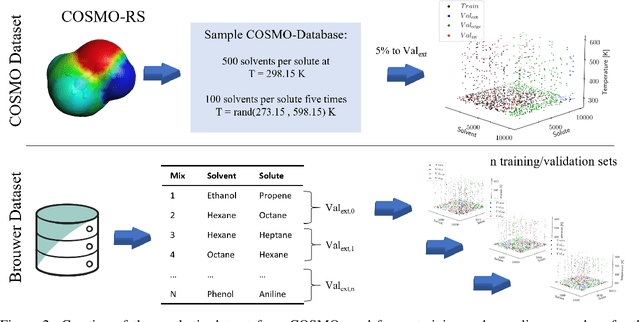
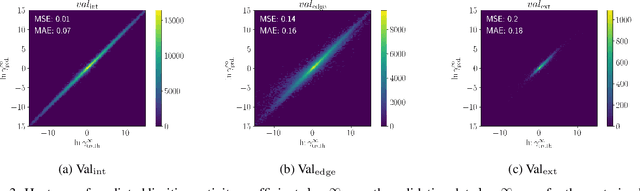
Knowledge of mixtures' phase equilibria is crucial in nature and technical chemistry. Phase equilibria calculations of mixtures require activity coefficients. However, experimental data on activity coefficients is often limited due to high cost of experiments. For an accurate and efficient prediction of activity coefficients, machine learning approaches have been recently developed. However, current machine learning approaches still extrapolate poorly for activity coefficients of unknown molecules. In this work, we introduce the SMILES-to-Properties-Transformer (SPT), a natural language processing network to predict binary limiting activity coefficients from SMILES codes. To overcome the limitations of available experimental data, we initially train our network on a large dataset of synthetic data sampled from COSMO-RS (10 Million data points) and then fine-tune the model on experimental data (20 870 data points). This training strategy enables SPT to accurately predict limiting activity coefficients even for unknown molecules, cutting the mean prediction error in half compared to state-of-the-art models for activity coefficient predictions such as COSMO-RS, UNIFAC, and improving on recent machine learning approaches.