 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient and Accurate Spatial Mixing of Machine Learned Interatomic Potentials for Materials Science
Feb 26, 2025

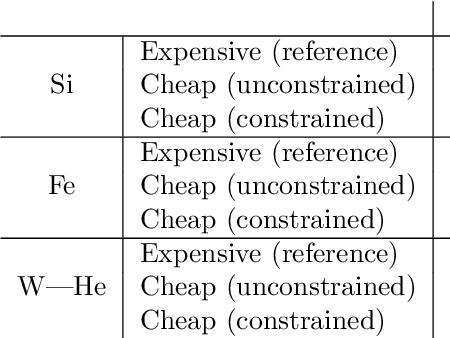
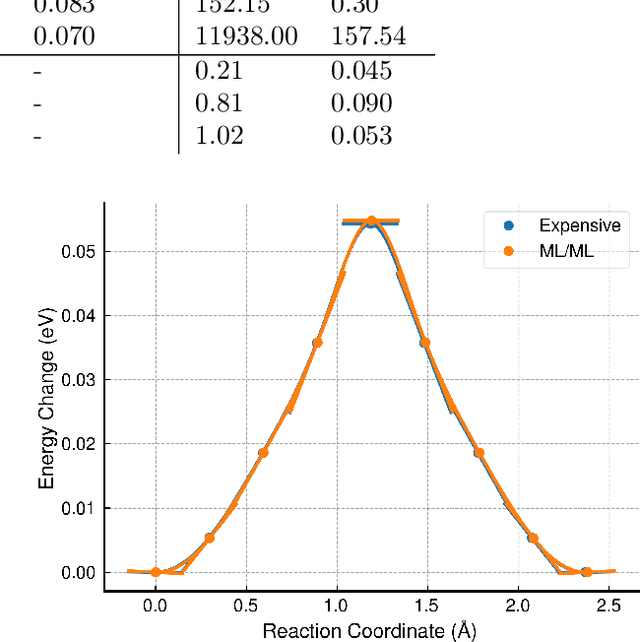
Machine-learned interatomic potentials offer near first-principles accuracy but are computationally expensive, limiting their application in large-scale molecular dynamics simulations. Inspired by quantum mechanics/molecular mechanics methods, we present ML-MIX, an efficient and flexible LAMMPS package for accelerating simulations by spatially mixing interatomic potentials of different complexities. Through constrained linear fitting, we show it is possible to generate a 'cheap' approximate model which closely matches an 'expensive' reference in relevant regions of configuration space. We demonstrate the capability of ML-MIX through case-studies in Si, Fe, and W-He systems, achieving up to an 11x speedup on 8,000 atom systems without sacrificing accuracy on static and dynamic quantities, including calculation of minimum energy paths and dynamical simulations of defect diffusion. For larger domain sizes, we show that the achievable speedup of ML-MIX simulations is limited only by the relative speed of the cheap potential over the expensive potential. The ease of use and flexible nature of this method will extend the practical reach of MLIPs throughout computational materials science, enabling parsimonious application to large spatial and temporal domains.
Misspecification uncertainties in near-deterministic regression
Feb 02, 2024The expected loss is an upper bound to the model generalization error which admits robust PAC-Bayes bounds for learning. However, loss minimization is known to ignore misspecification, where models cannot exactly reproduce observations. This leads to significant underestimates of parameter uncertainties in the large data, or underparameterized, limit. We analyze the generalization error of near-deterministic, misspecified and underparametrized surrogate models, a regime of broad relevance in science and engineering. We show posterior distributions must cover every training point to avoid a divergent generalization error and derive an ensemble {ansatz} that respects this constraint, which for linear models incurs minimal overhead. The efficient approach is demonstrated on model problems before application to high dimensional datasets in atomistic machine learning. Parameter uncertainties from misspecification survive in the underparametrized limit, giving accurate prediction and bounding of test errors.