 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient and Accurate Spatial Mixing of Machine Learned Interatomic Potentials for Materials Science
Feb 26, 2025

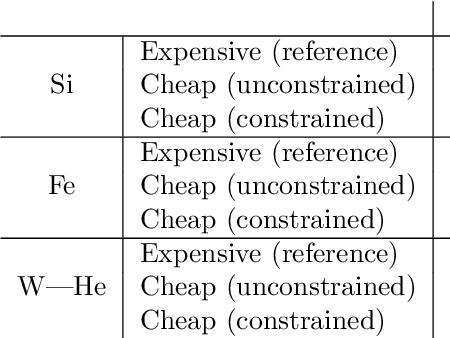
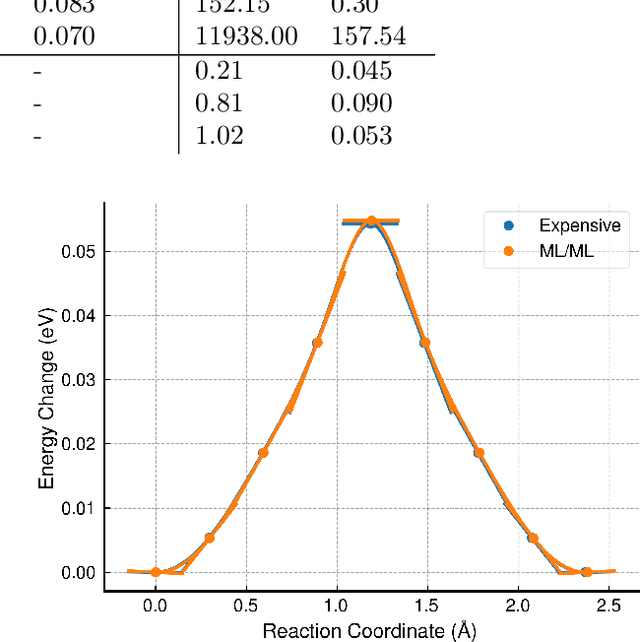
Machine-learned interatomic potentials offer near first-principles accuracy but are computationally expensive, limiting their application in large-scale molecular dynamics simulations. Inspired by quantum mechanics/molecular mechanics methods, we present ML-MIX, an efficient and flexible LAMMPS package for accelerating simulations by spatially mixing interatomic potentials of different complexities. Through constrained linear fitting, we show it is possible to generate a 'cheap' approximate model which closely matches an 'expensive' reference in relevant regions of configuration space. We demonstrate the capability of ML-MIX through case-studies in Si, Fe, and W-He systems, achieving up to an 11x speedup on 8,000 atom systems without sacrificing accuracy on static and dynamic quantities, including calculation of minimum energy paths and dynamical simulations of defect diffusion. For larger domain sizes, we show that the achievable speedup of ML-MIX simulations is limited only by the relative speed of the cheap potential over the expensive potential. The ease of use and flexible nature of this method will extend the practical reach of MLIPs throughout computational materials science, enabling parsimonious application to large spatial and temporal domains.
Accelerating a hybrid continuum-atomistic fluidic model with on-the-fly machine learning
Mar 15, 2016



We present a hybrid continuum-atomistic scheme which combines molecular dynamics (MD) simulations with on-the-fly machine learning techniques for the accurate and efficient prediction of multiscale fluidic systems. By using a Gaussian process as a surrogate model for the computationally expensive MD simulations, we use Bayesian inference to predict the system behaviour at the atomistic scale, purely by consideration of the macroscopic inputs and outputs. Whenever the uncertainty of this prediction is greater than a predetermined acceptable threshold, a new MD simulation is performed to continually augment the database, which is never required to be complete. This provides a substantial enhancement to the current generation of hybrid methods, which often require many similar atomistic simulations to be performed, discarding information after it is used once. We apply our hybrid scheme to nano-confined unsteady flow through a high-aspect-ratio converging-diverging channel, and make comparisons between the new scheme and full MD simulations for a range of uncertainty thresholds and initial databases. For low thresholds, our hybrid solution is highly accurate\,---\,within the thermal noise of a full MD simulation. As the uncertainty threshold is raised, the accuracy of our scheme decreases and the computational speed-up increases (relative to a full MD simulation), enabling the compromise between precision and efficiency to be tuned. The speed-up of our hybrid solution ranges from an order of magnitude, with no initial database, to cases where an extensive initial database ensures no new MD simulations are required.