 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomating reward function configuration for drug design
Dec 15, 2023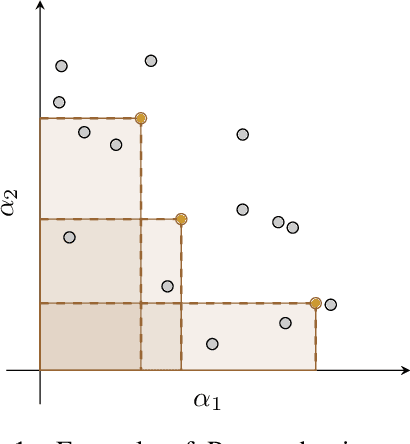
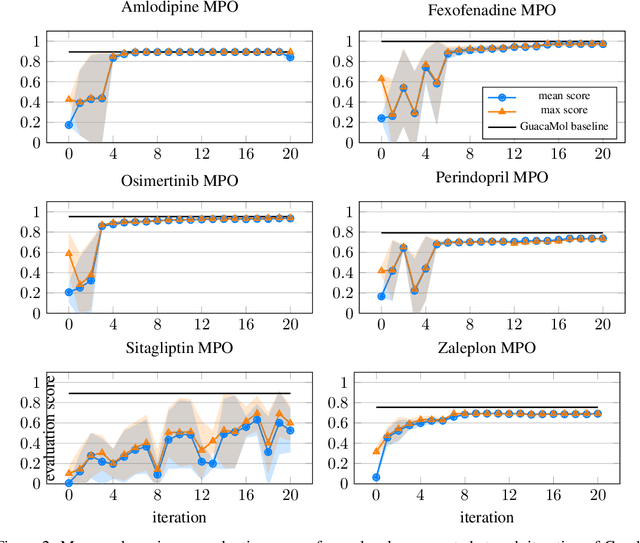
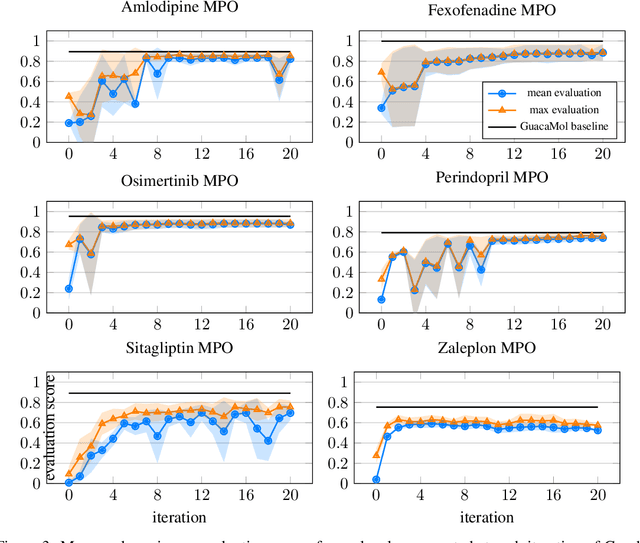
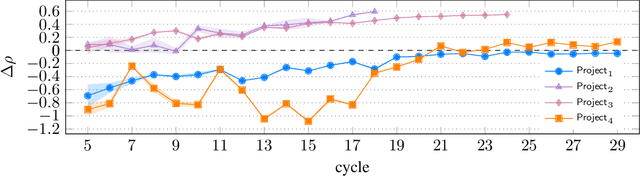
Designing reward functions that guide generative molecular design (GMD) algorithms to desirable areas of chemical space is of critical importance in AI-driven drug discovery. Traditionally, this has been a manual and error-prone task; the selection of appropriate computational methods to approximate biological assays is challenging and the aggregation of computed values into a single score even more so, leading to potential reliance on trial-and-error approaches. We propose a novel approach for automated reward configuration that relies solely on experimental data, mitigating the challenges of manual reward adjustment on drug discovery projects. Our method achieves this by constructing a ranking over experimental data based on Pareto dominance over the multi-objective space, then training a neural network to approximate the reward function such that rankings determined by the predicted reward correlate with those determined by the Pareto dominance relation. We validate our method using two case studies. In the first study we simulate Design-Make-Test-Analyse (DMTA) cycles by alternating reward function updates and generative runs guided by that function. We show that the learned function adapts over time to yield compounds that score highly with respect to evaluation functions taken from the literature. In the second study we apply our algorithm to historical data from four real drug discovery projects. We show that our algorithm yields reward functions that outperform the predictive accuracy of human-defined functions, achieving an improvement of up to 0.4 in Spearman's correlation against a ground truth evaluation function that encodes the target drug profile for that project. Our method provides an efficient data-driven way to configure reward functions for GMD, and serves as a strong baseline for future research into transformative approaches for the automation of drug discovery.