 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCUDA optimized Neural Network predicts blood glucose control from quantified joint mobility and anthropometrics
Aug 19, 2019
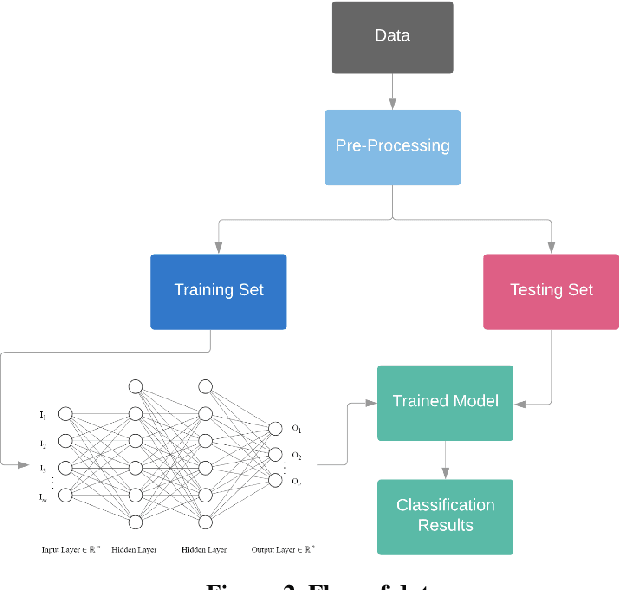
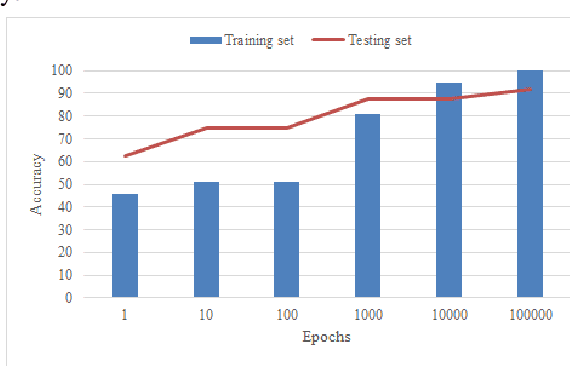

Neural network training entails heavy computation with obvious bottlenecks. The Compute Unified Device Architecture (CUDA) programming model allows us to accelerate computation by passing the processing workload from the CPU to the graphics processing unit (GPU). In this paper, we leveraged the power of Nvidia GPUs to parallelize all of the computation involved in training, to accelerate a backpropagation feed-forward neural network with one hidden layer using CUDA and C++. This optimized neural network was tasked with predicting the level of glycated hemoglobin (HbA1c) from non-invasive markers. The rate of increase in the prevalence of Diabetes Mellitus has resulted in an urgent need for early detection and accurate diagnosis. However, due to the invasiveness and limitations of conventional tests, alternate means are being considered. Limited Joint Mobility (LJM) has been reported as an indicator for poor glycemic control. LJM of the fingers is quantified and its link to HbA1c is investigated along with other potential non-invasive markers of HbA1c. We collected readings of 33 potential markers from 120 participants at a clinic in south Trinidad. Our neural network achieved 95.65% accuracy on the training and 86.67% accuracy on the testing set for male participants and 97.73% and 66.67% accuracy on the training and testing sets for female participants. Using 960 CUDA cores from a Nvidia GeForce GTX 660, our parallelized neural network was trained 50 times faster on both subsets, than its corresponding CPU implementation on an Intel Core (TM) i7-3630QM 2.40 GHz CPU.
The efficacy of various machine learning models for multi-class classification of RNA-seq expression data
Aug 19, 2019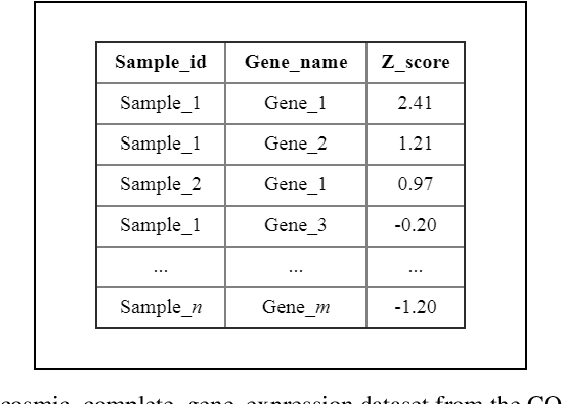


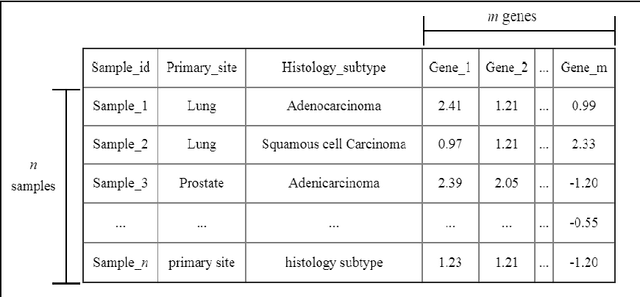
Late diagnosis and high costs are key factors that negatively impact the care of cancer patients worldwide. Although the availability of biological markers for the diagnosis of cancer type is increasing, costs and reliability of tests currently present a barrier to the adoption of their routine use. There is a pressing need for accurate methods that enable early diagnosis and cover a broad range of cancers. The use of machine learning and RNA-seq expression analysis has shown promise in the classification of cancer type. However, research is inconclusive about which type of machine learning models are optimal. The suitability of five algorithms were assessed for the classification of 17 different cancer types. Each algorithm was fine-tuned and trained on the full array of 18,015 genes per sample, for 4,221 samples (75 % of the dataset). They were then tested with 1,408 samples (25 % of the dataset) for which cancer types were withheld to determine the accuracy of prediction. The results show that ensemble algorithms achieve 100% accuracy in the classification of 14 out of 17 types of cancer. The clustering and classification models, while faster than the ensembles, performed poorly due to the high level of noise in the dataset. When the features were reduced to a list of 20 genes, the ensemble algorithms maintained an accuracy above 95% as opposed to the clustering and classification models.