 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHybrid Quantum--Classical Machine Learning Potential with Variational Quantum Circuits
Aug 06, 2025
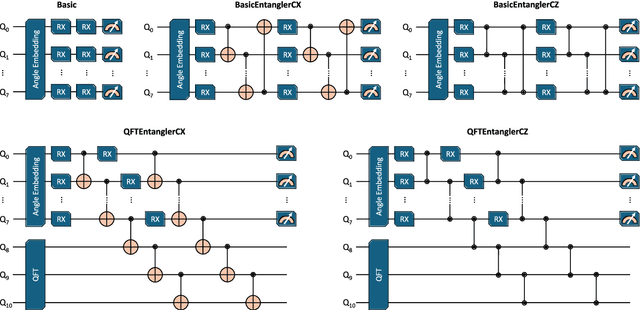


Quantum algorithms for simulating large and complex molecular systems are still in their infancy, and surpassing state-of-the-art classical techniques remains an ever-receding goal post. A promising avenue of inquiry in the meanwhile is to seek practical advantages through hybrid quantum-classical algorithms, which combine conventional neural networks with variational quantum circuits (VQCs) running on today's noisy intermediate-scale quantum (NISQ) hardware. Such hybrids are well suited to NISQ hardware. The classical processor performs the bulk of the computation, while the quantum processor executes targeted sub-tasks that supply additional non-linearity and expressivity. Here, we benchmark a purely classical E(3)-equivariant message-passing machine learning potential (MLP) against a hybrid quantum-classical MLP for predicting density functional theory (DFT) properties of liquid silicon. In our hybrid architecture, every readout in the message-passing layers is replaced by a VQC. Molecular dynamics simulations driven by the HQC-MLP reveal that an accurate reproduction of high-temperature structural and thermodynamic properties is achieved with VQCs. These findings demonstrate a concrete scenario in which NISQ-compatible HQC algorithm could deliver a measurable benefit over the best available classical alternative, suggesting a viable pathway toward near-term quantum advantage in materials modeling.
Machine Learning Nonadiabatic Dynamics: Eliminating Phase Freedom of Nonadiabatic Couplings with the State-Intraction State-Averaged Spin-Restricted Ensemble-Referenced Kohn-Sham Approach
Oct 30, 2024Excited-state molecular dynamics (ESMD) simulations near conical intersections (CIs) pose significant challenges when using machine learning potentials (MLPs). Although MLPs have gained recognition for their integration into mixed quantum-classical (MQC) methods, such as trajectory surface hopping (TSH), and their capacity to model correlated electron-nuclear dynamics efficiently, difficulties persist in managing nonadiabatic dynamics. Specifically, singularities at CIs and double-valued coupling elements result in discontinuities that disrupt the smoothness of predictive functions. Partial solutions have been provided by learning diabatic Hamiltonians with phaseless loss functions to these challenges. However, a definitive method for addressing the discontinuities caused by CIs and double-valued coupling elements has yet to be developed. Here, we introduce the phaseless coupling term, $\Delta^2$, derived from the square of the off-diagonal elements of the diabatic Hamiltonian in the SSR(2,2) formalism. This approach improves the stability and accuracy of the MLP model by addressing the issues arising from CI singularities and double-valued coupling functions. We apply this method to the penta-2,4-dieniminium cation (PSB3), demonstrating its effectiveness in improving MLP training for ML-based nonadiabatic dynamics. Our results show that the $\Delta^2$ based ML-ESMD method can reproduce ab initio ESMD simulations, underscoring its potential and efficiency for broader applications, particularly in large-scale and long-timescale ESMD simulations.
A Bayesian Committee Machine Potential for Oxygen-containing Organic Compounds
Mar 02, 2024



Understanding the pivotal role of oxygen-containing organic compounds in serving as an energy source for living organisms and contributing to protein formation is crucial in the field of biochemistry. This study addresses the challenge of comprehending protein-protein interactions (PPI) and developing predicitive models for proteins and organic compounds, with a specific focus on quantifying their binding affinity. Here, we introduce the active Bayesian Committee Machine (BCM) potential, specifically designed to predict oxygen-containing organic compounds within eight groups of CHO. The BCM potential adopts a committee-based approach to tackle scalability issues associated with kernel regressors, particularly when dealing with large datasets. Its adaptable structure allows for efficient and cost-effective expansion, maintaing both transferability and scalability. Through systematic benchmarking, we position the sparse BCM potential as a promising contender in the pursuit of a universal machine learning potential.
Learned Mappings for Targeted Free Energy Perturbation between Peptide Conformations
Jun 24, 2023Targeted free energy perturbation uses an invertible mapping to promote configuration space overlap and the convergence of free energy estimates. However, developing suitable mappings can be challenging. Wirnsberger et al. (2020) demonstrated the use of machine learning to train deep neural networks that map between Boltzmann distributions for different thermodynamic states. Here, we adapt their approach to free energy differences of a flexible bonded molecule, deca-alanine, with harmonic biases with different spring centers. When the neural network is trained until ``early stopping'' - when the loss value of the test set increases - we calculate accurate free energy differences between thermodynamic states with spring centers separated by 1 \r{A} and sometimes 2 \r{A}. For more distant thermodynamic states, the mapping does not produce structures representative of the target state and the method does not reproduce reference calculations.