 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting Oxide Glass Properties with Low Complexity Neural Network and Physical and Chemical Descriptors
Oct 19, 2022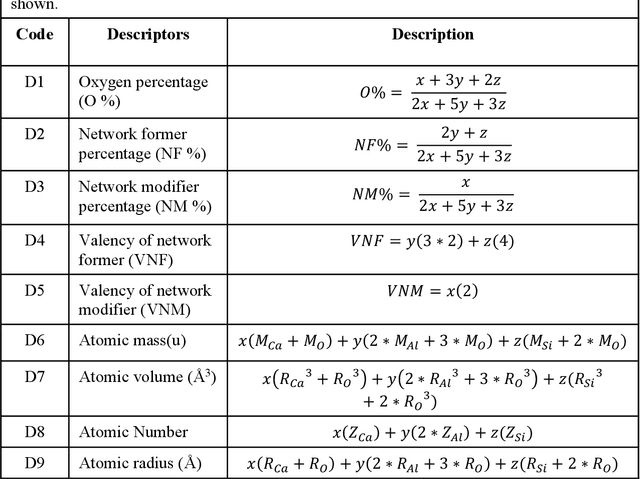
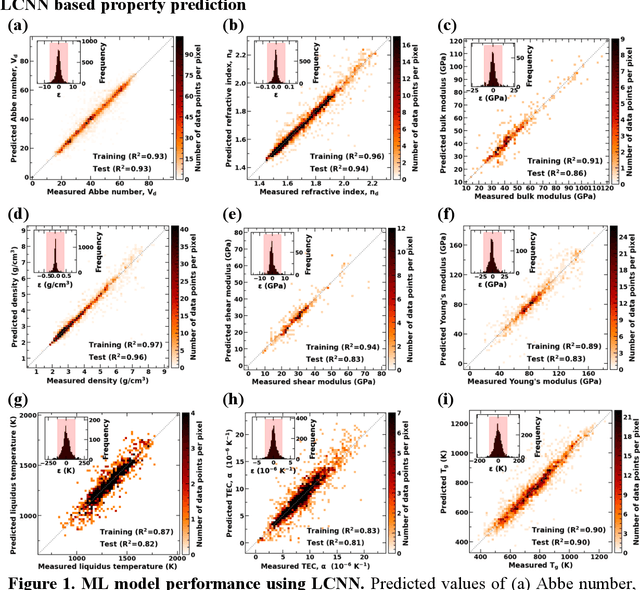
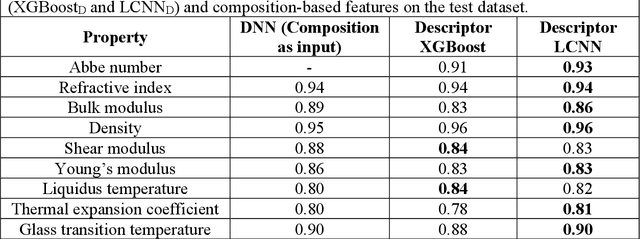
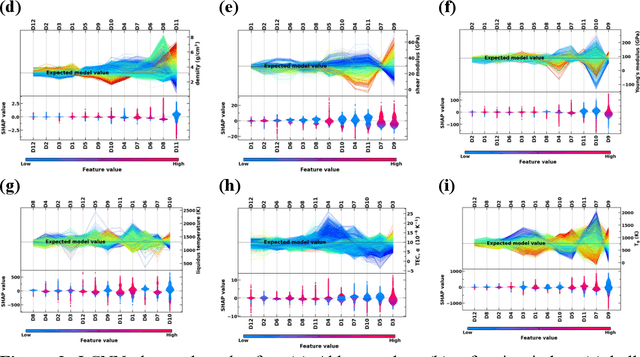
Due to their disordered structure, glasses present a unique challenge in predicting the composition-property relationships. Recently, several attempts have been made to predict the glass properties using machine learning techniques. However, these techniques have the limitations, namely, (i) predictions are limited to the components that are present in the original dataset, and (ii) predictions towards the extreme values of the properties, important regions for new materials discovery, are not very reliable due to the sparse datapoints in this region. To address these challenges, here we present a low complexity neural network (LCNN) that provides improved performance in predicting the properties of oxide glasses. In addition, we combine the LCNN with physical and chemical descriptors that allow the development of universal models that can provide predictions for components beyond the training set. By training on a large dataset (~50000) of glass components, we show the LCNN outperforms state-of-the-art algorithms such as XGBoost. In addition, we interpret the LCNN models using Shapely additive explanations to gain insights into the role played by the descriptors in governing the property. Finally, we demonstrate the universality of the LCNN models by predicting the properties for glasses with new components that were not present in the original training set. Altogether, the present approach provides a promising direction towards accelerated discovery of novel glass compositions.