 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePartition Decomposition for Roll Call Data
Aug 13, 2011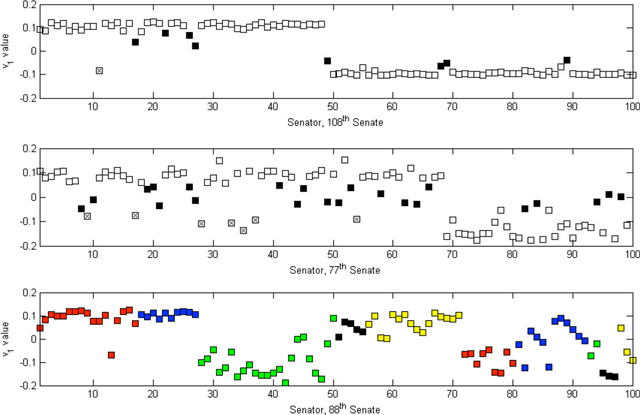
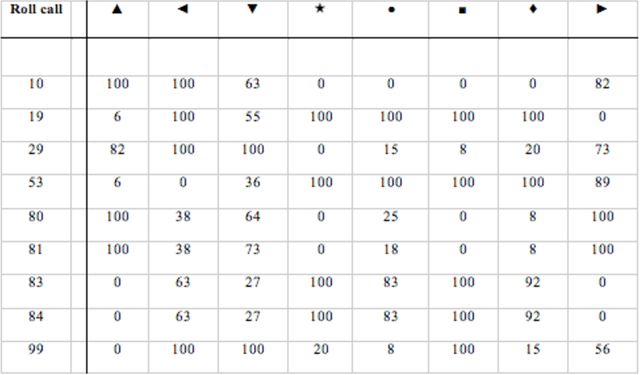

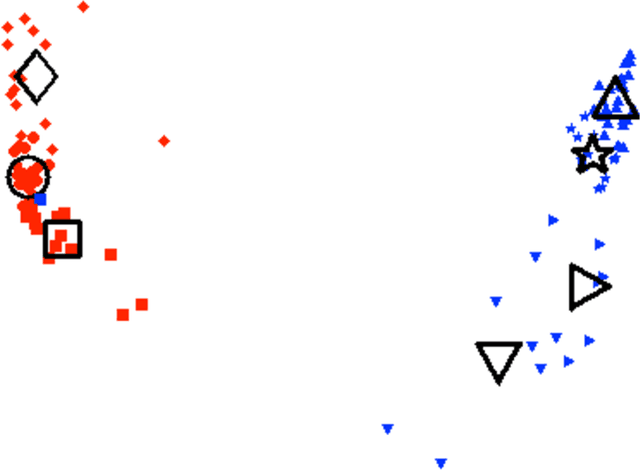
In this paper we bring to bear some new tools from statistical learning on the analysis of roll call data. We present a new data-driven model for roll call voting that is geometric in nature. We construct the model by adapting the "Partition Decoupling Method," an unsupervised learning technique originally developed for the analysis of families of time series, to produce a multiscale geometric description of a weighted network associated to a set of roll call votes. Central to this approach is the quantitative notion of a "motivation," a cluster-based and learned basis element that serves as a building block in the representation of roll call data. Motivations enable the formulation of a quantitative description of ideology and their data-dependent nature makes possible a quantitative analysis of the evolution of ideological factors. This approach is generally applicable to roll call data and we apply it in particular to the historical roll call voting of the U.S. House and Senate. This methodology provides a mechanism for estimating the dimension of the underlying action space. We determine that the dominant factors form a low- (one- or two-) dimensional representation with secondary factors adding higher-dimensional features. In this way our work supports and extends the findings of both Poole-Rosenthal and Heckman-Snyder concerning the dimensionality of the action space. We give a detailed analysis of several individual Senates and use the AdaBoost technique from statistical learning to determine those votes with the most powerful discriminatory value. When used as a predictive model, this geometric view significantly outperforms spatial models such as the Poole-Rosenthal DW-NOMINATE model and the Heckman-Snyder 6-factor model, both in raw accuracy as well as Aggregate Proportional Reduced Error (APRE).
Partition Decoupling for Multi-gene Analysis of Gene Expression Profiling Data
Mar 17, 2011



We present the extention and application of a new unsupervised statistical learning technique--the Partition Decoupling Method--to gene expression data. Because it has the ability to reveal non-linear and non-convex geometries present in the data, the PDM is an improvement over typical gene expression analysis algorithms, permitting a multi-gene analysis that can reveal phenotypic differences even when the individual genes do not exhibit differential expression. Here, we apply the PDM to publicly-available gene expression data sets, and demonstrate that we are able to identify cell types and treatments with higher accuracy than is obtained through other approaches. By applying it in a pathway-by-pathway fashion, we demonstrate how the PDM may be used to find sets of mechanistically-related genes that discriminate phenotypes.