 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFragmentFlow: Scalable Transition State Generation for Large Molecules
Feb 02, 2026Transition states (TSs) are central to understanding and quantitatively predicting chemical reactivity and reaction mechanisms. Although traditional TS generation methods are computationally expensive, recent generative modeling approaches have enabled chemically meaningful TS prediction for relatively small molecules. However, these methods fail to generalize to practically relevant reaction substrates because of distribution shifts induced by increasing molecular sizes. Furthermore, TS geometries for larger molecules are not available at scale, making it infeasible to train generative models from scratch on such molecules. To address these challenges, we introduce FragmentFlow: a divide-and-conquer approach that trains a generative model to predict TS geometries for the reactive core atoms, which define the reaction mechanism. The full TS structure is then reconstructed by re-attaching substituent fragments to the predicted core. By operating on reactive cores, whose size and composition remain relatively invariant across molecular contexts, FragmentFlow mitigates distribution shifts in generative modeling. Evaluated on a new curated dataset of reactions involving reactants with up to 33 heavy atoms, FragmentFlow correctly identifies 90% of TSs while requiring 30% fewer saddle-point optimization steps than classical initialization schemes. These results point toward scalable TS generation for high-throughput reactivity studies.
Substrate Scope Contrastive Learning: Repurposing Human Bias to Learn Atomic Representations
Feb 19, 2024

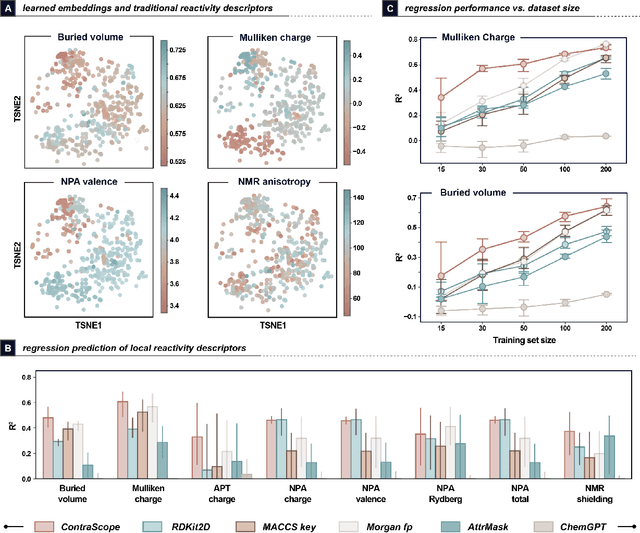
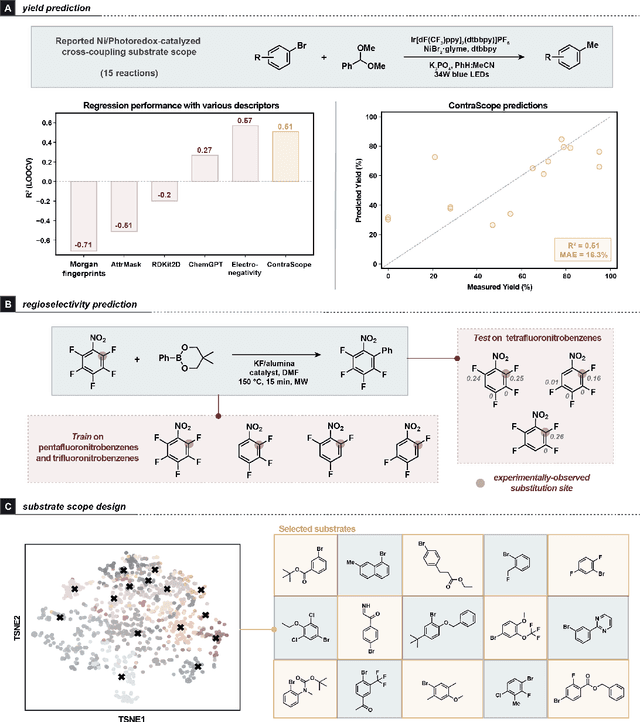
Learning molecular representation is a critical step in molecular machine learning that significantly influences modeling success, particularly in data-scarce situations. The concept of broadly pre-training neural networks has advanced fields such as computer vision, natural language processing, and protein engineering. However, similar approaches for small organic molecules have not achieved comparable success. In this work, we introduce a novel pre-training strategy, substrate scope contrastive learning, which learns atomic representations tailored to chemical reactivity. This method considers the grouping of substrates and their yields in published substrate scope tables as a measure of their similarity or dissimilarity in terms of chemical reactivity. We focus on 20,798 aryl halides in the CAS Content Collection spanning thousands of publications to learn a representation of aryl halide reactivity. We validate our pre-training approach through both intuitive visualizations and comparisons to traditional reactivity descriptors and physical organic chemistry principles. The versatility of these embeddings is further evidenced in their application to yield prediction, regioselectivity prediction, and the diverse selection of new substrates. This work not only presents a chemistry-tailored neural network pre-training strategy to learn reactivity-aligned atomic representations, but also marks a first-of-its-kind approach to benefit from the human bias in substrate scope design.