 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMany-body Expansion Based Machine Learning Models for Octahedral Transition Metal Complexes
Oct 12, 2024Graph-based machine learning models for materials properties show great potential to accelerate virtual high-throughput screening of large chemical spaces. However, in their simplest forms, graph-based models do not include any 3D information and are unable to distinguish stereoisomers such as those arising from different orderings of ligands around a metal center in coordination complexes. In this work we present a modification to revised autocorrelation descriptors, our molecular graph featurization method for machine learning various spin state dependent properties of octahedral transition metal complexes (TMCs). Inspired by analytical semi-empirical models for TMCs, the new modeling strategy is based on the many-body expansion (MBE) and allows one to tune the captured stereoisomer information by changing the truncation order of the MBE. We present the necessary modifications to include this approach in two commonly used machine learning methods, kernel ridge regression and feed-forward neural networks. On a test set composed of all possible isomers of binary transition metal complexes, the best MBE models achieve mean absolute errors of 2.75 kcal/mol on spin-splitting energies and 0.26 eV on frontier orbital energy gaps, a 30-40% reduction in error compared to models based on our previous approach. We also observe improved generalization to previously unseen ligands where the best-performing models exhibit mean absolute errors of 4.00 kcal/mol (i.e., a 0.73 kcal/mol reduction) on the spin-splitting energies and 0.53 eV (i.e., a 0.10 eV reduction) on the frontier orbital energy gaps. Because the new approach incorporates insights from electronic structure theory, such as ligand additivity relationships, these models exhibit systematic generalization from homoleptic to heteroleptic complexes, allowing for efficient screening of TMC search spaces.
A Transferable Recommender Approach for Selecting the Best Density Functional Approximations in Chemical Discovery
Jul 21, 2022

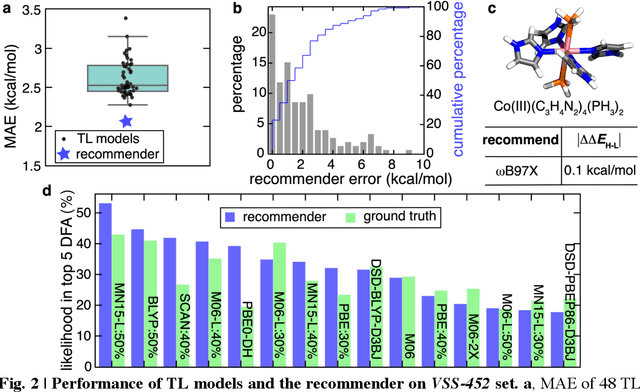

Approximate density functional theory (DFT) has become indispensable owing to its cost-accuracy trade-off in comparison to more computationally demanding but accurate correlated wavefunction theory. To date, however, no single density functional approximation (DFA) with universal accuracy has been identified, leading to uncertainty in the quality of data generated from DFT. With electron density fitting and transfer learning, we build a DFA recommender that selects the DFA with the lowest expected error with respect to gold standard but cost-prohibitive coupled cluster theory in a system-specific manner. We demonstrate this recommender approach on vertical spin-splitting energy evaluation for challenging transition metal complexes. Our recommender predicts top-performing DFAs and yields excellent accuracy (ca. 2 kcal/mol) for chemical discovery, outperforming both individual transfer learning models and the single best functional in a set of 48 DFAs. We demonstrate the transferability of the DFA recommender to experimentally synthesized compounds with distinct chemistry.