 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeep Learning and Knowledge-Based Methods for Computer Aided Molecular Design -- Toward a Unified Approach: State-of-the-Art and Future Directions
May 18, 2020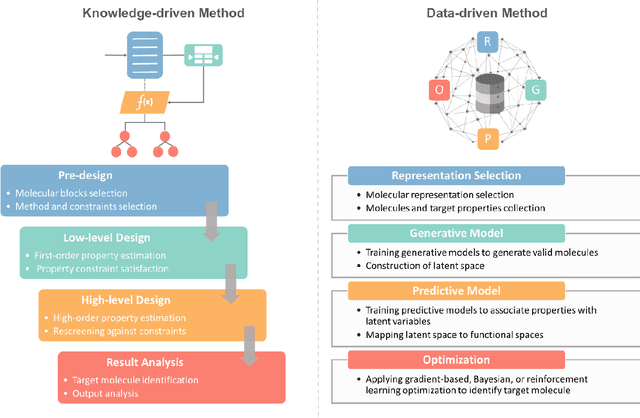
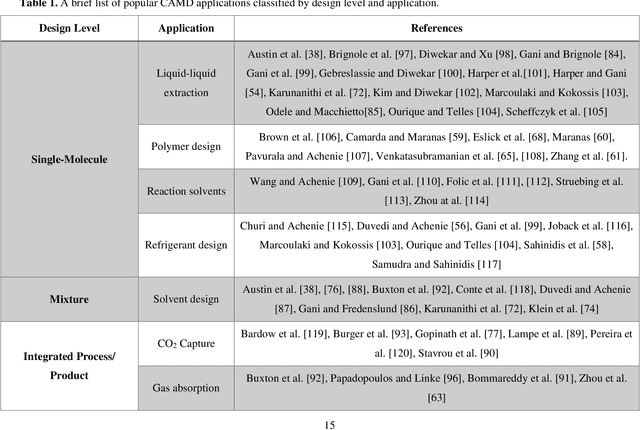
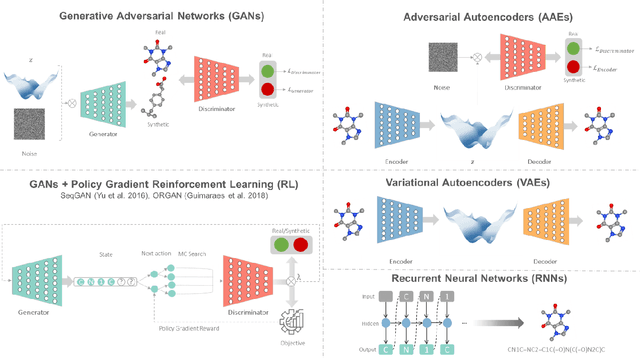
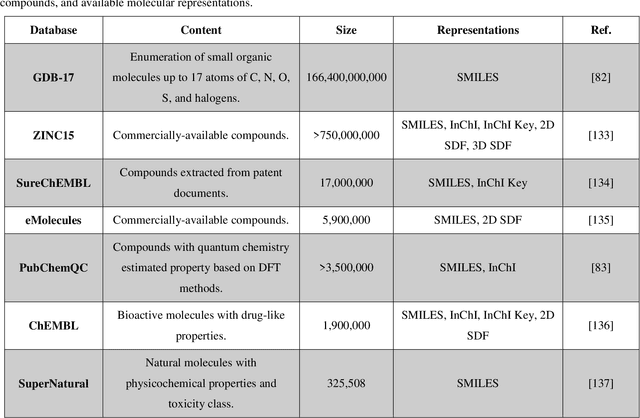
The optimal design of compounds through manipulating properties at the molecular level is often the key to considerable scientific advances and improved process systems performance. This paper highlights key trends, challenges, and opportunities underpinning the Computer-Aided Molecular Design (CAMD) problems. A brief review of knowledge-driven property estimation methods and solution techniques, as well as corresponding CAMD tools and applications, are first presented. In view of the computational challenges plaguing knowledge-based methods and techniques, we survey the current state-of-the-art applications of deep learning to molecular design as a fertile approach towards overcoming computational limitations and navigating uncharted territories of the chemical space. The main focus of the survey is given to deep generative modeling of molecules under various deep learning architectures and different molecular representations. Further, the importance of benchmarking and empirical rigor in building deep learning models is spotlighted. The review article also presents a detailed discussion of the current perspectives and challenges of knowledge-based and data-driven CAMD and identifies key areas for future research directions. Special emphasis is on the fertile avenue of hybrid modeling paradigm, in which deep learning approaches are exploited while leveraging the accumulated wealth of knowledge-driven CAMD methods and tools.