 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtofold II: Enhanced Model and Implementation for Kinetostatic Protein Folding
Nov 14, 2017


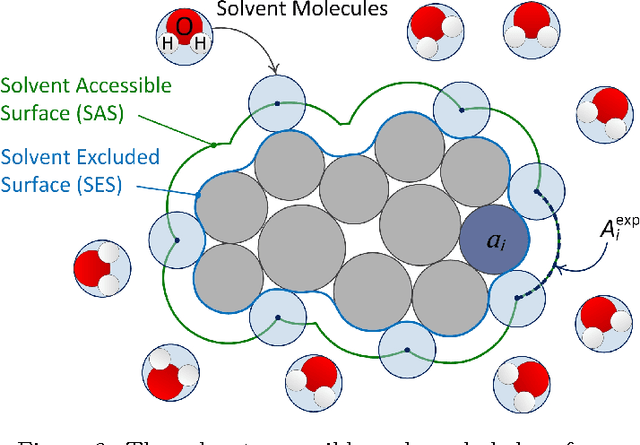
A reliable prediction of 3D protein structures from sequence data remains a big challenge due to both theoretical and computational difficulties. We have previously shown that our kinetostatic compliance method (KCM) implemented into the Protofold package can overcome some of the key difficulties faced by other de novo structure prediction methods, such as the very small time steps required by the molecular dynamics (MD) approaches or the very large number of samples needed by the Monte Carlo (MC) sampling techniques. In this article, we improve the free energy formulation used in Protofold by including the typically underrated entropic effects, imparted due to differences in hydrophobicity of the chemical groups, which dominate the folding of most water-soluble proteins. In addition to the model enhancement, we revisit the numerical implementation by redesigning the algorithms and introducing efficient data structures that reduce the expected complexity from quadratic to linear. Moreover, we develop and optimize parallel implementations of the algorithms on both central and graphics processing units (CPU/GPU) achieving speed-ups up to two orders of magnitude on the GPU. Our simulations are consistent with the general behavior observed in the folding process in aqueous solvent, confirming the effectiveness of model improvements. We report on the folding process at multiple levels; namely, the formation of secondary structural elements and tertiary interactions between secondary elements or across larger domains. We also observe significant enhancements in running times that make the folding simulation tractable for large molecules.
* Shorter versions were presented in two conference papers in ASME International Design Engineering Technical Conferences (IDETC'2013)