 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCombining Cost-Sensitive Classification with Negative Selection for Protein Function Prediction
May 18, 2018
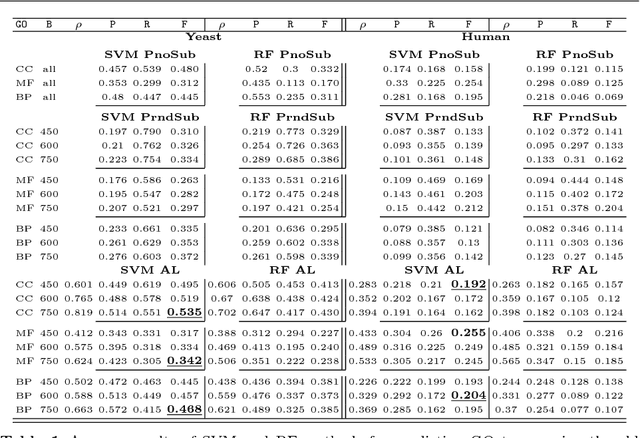
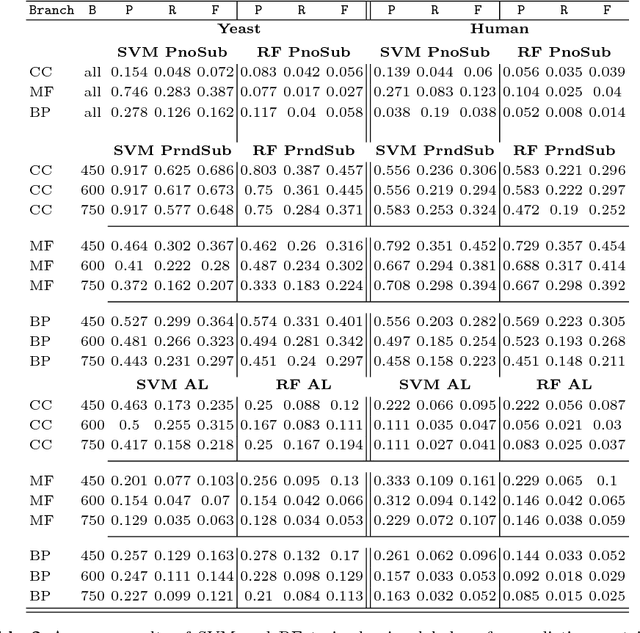
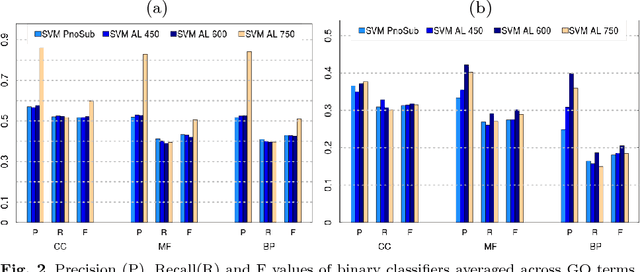
Motivation: Computational methods play a central role in annotating the functions of large amounts of proteins delivered by high-throughput technologies. Despite the encouraging results achieved by these methods, many functions still have a very low number of verified protein annotations, leading to a pronounced imbalance between annotated and unannotated proteins. Furthermore, functional taxonomies rarely report negative annotations. This leaves ill defined the set of negative examples, which is crucial for training the majority of machine learning methods. In practice, neglecting data imbalance and the problem of selecing negative examples can strongly limit the accuracy of protein function prediction. Results: We present a novel approach combining a suitable imbalance-aware classification strategy, addressing the scarcity of annotated proteins, with an active learning strategy for selecting the most reliable negative examples. When implemented in a Support Vector Machine, this combined approach shows improved accuracy on yeast and human proteomes over standard SVM and top-performing function prediction tools
Multitask Protein Function Prediction Through Task Dissimilarity
Nov 03, 2016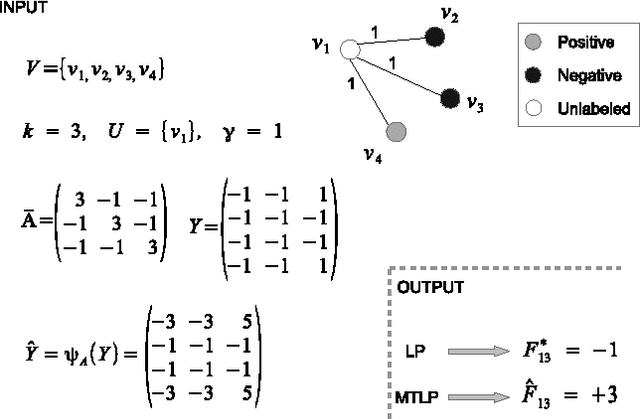
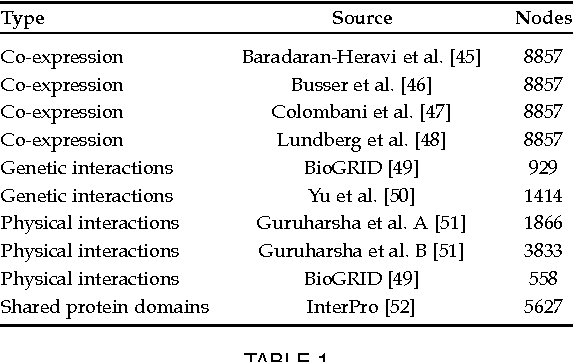
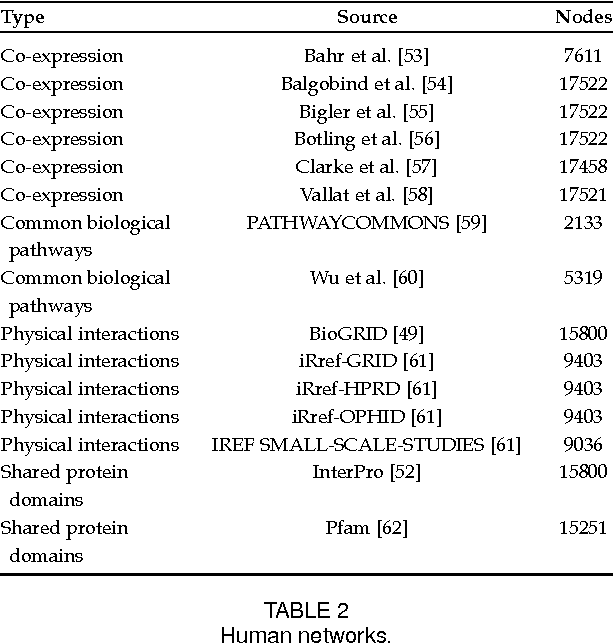

Automated protein function prediction is a challenging problem with distinctive features, such as the hierarchical organization of protein functions and the scarcity of annotated proteins for most biological functions. We propose a multitask learning algorithm addressing both issues. Unlike standard multitask algorithms, which use task (protein functions) similarity information as a bias to speed up learning, we show that dissimilarity information enforces separation of rare class labels from frequent class labels, and for this reason is better suited for solving unbalanced protein function prediction problems. We support our claim by showing that a multitask extension of the label propagation algorithm empirically works best when the task relatedness information is represented using a dissimilarity matrix as opposed to a similarity matrix. Moreover, the experimental comparison carried out on three model organism shows that our method has a more stable performance in both "protein-centric" and "function-centric" evaluation settings.