 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeG-RAG: Knowledge Expansion in Material Science
Nov 21, 2024

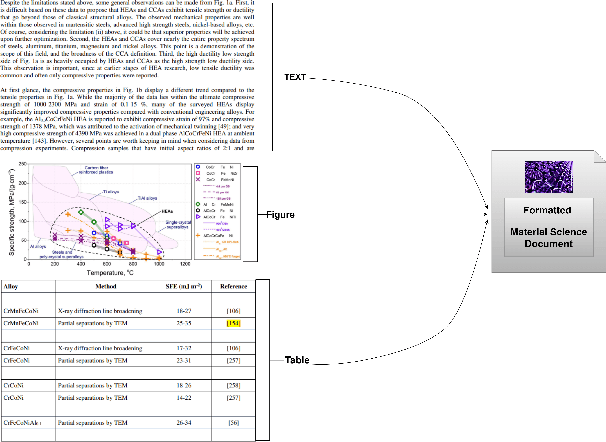

In the field of Material Science, effective information retrieval systems are essential for facilitating research. Traditional Retrieval-Augmented Generation (RAG) approaches in Large Language Models (LLMs) often encounter challenges such as outdated information, hallucinations, limited interpretability due to context constraints, and inaccurate retrieval. To address these issues, Graph RAG integrates graph databases to enhance the retrieval process. Our proposed method processes Material Science documents by extracting key entities (referred to as MatIDs) from sentences, which are then utilized to query external Wikipedia knowledge bases (KBs) for additional relevant information. We implement an agent-based parsing technique to achieve a more detailed representation of the documents. Our improved version of Graph RAG called G-RAG further leverages a graph database to capture relationships between these entities, improving both retrieval accuracy and contextual understanding. This enhanced approach demonstrates significant improvements in performance for domains that require precise information retrieval, such as Material Science.
Graph neural network framework for energy mapping of hybrid monte-carlo molecular dynamics simulations of Medium Entropy Alloys
Nov 20, 2024



Machine learning (ML) methods have drawn significant interest in material design and discovery. Graph neural networks (GNNs), in particular, have demonstrated strong potential for predicting material properties. The present study proposes a graph-based representation for modeling medium-entropy alloys (MEAs). Hybrid Monte-Carlo molecular dynamics (MC/MD) simulations are employed to achieve thermally stable structures across various annealing temperatures in an MEA. These simulations generate dump files and potential energy labels, which are used to construct graph representations of the atomic configurations. Edges are created between each atom and its 12 nearest neighbors without incorporating explicit edge features. These graphs then serve as input for a Graph Convolutional Neural Network (GCNN) based ML model to predict the system's potential energy. The GCNN architecture effectively captures the local environment and chemical ordering within the MEA structure. The GCNN-based ML model demonstrates strong performance in predicting potential energy at different steps, showing satisfactory results on both the training data and unseen configurations. Our approach presents a graph-based modeling framework for MEAs and high-entropy alloys (HEAs), which effectively captures the local chemical order (LCO) within the alloy structure. This allows us to predict key material properties influenced by LCO in both MEAs and HEAs, providing deeper insights into how atomic-scale arrangements affect the properties of these alloys.