 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAdvDINO: Domain-Adversarial Self-Supervised Representation Learning for Spatial Proteomics
Aug 07, 2025Self-supervised learning (SSL) has emerged as a powerful approach for learning visual representations without manual annotations. However, the robustness of standard SSL methods to domain shift -- systematic differences across data sources -- remains uncertain, posing an especially critical challenge in biomedical imaging where batch effects can obscure true biological signals. We present AdvDINO, a domain-adversarial self-supervised learning framework that integrates a gradient reversal layer into the DINOv2 architecture to promote domain-invariant feature learning. Applied to a real-world cohort of six-channel multiplex immunofluorescence (mIF) whole slide images from non-small cell lung cancer patients, AdvDINO mitigates slide-specific biases to learn more robust and biologically meaningful representations than non-adversarial baselines. Across $>5.46$ million mIF image tiles, the model uncovers phenotype clusters with distinct proteomic profiles and prognostic significance, and improves survival prediction in attention-based multiple instance learning. While demonstrated on mIF data, AdvDINO is broadly applicable to other imaging domains -- including radiology, remote sensing, and autonomous driving -- where domain shift and limited annotated data hinder model generalization and interpretability.
Fluoroformer: Scaling multiple instance learning to multiplexed images via attention-based channel fusion
Nov 13, 2024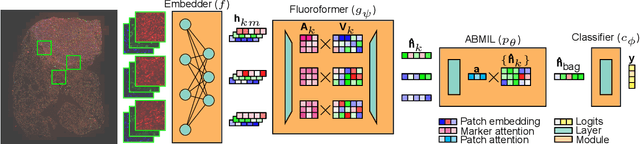
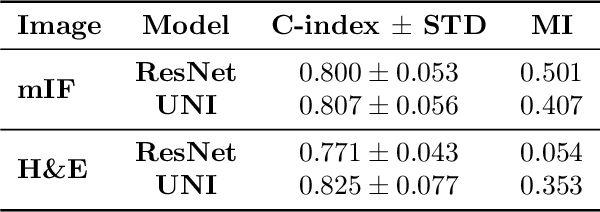

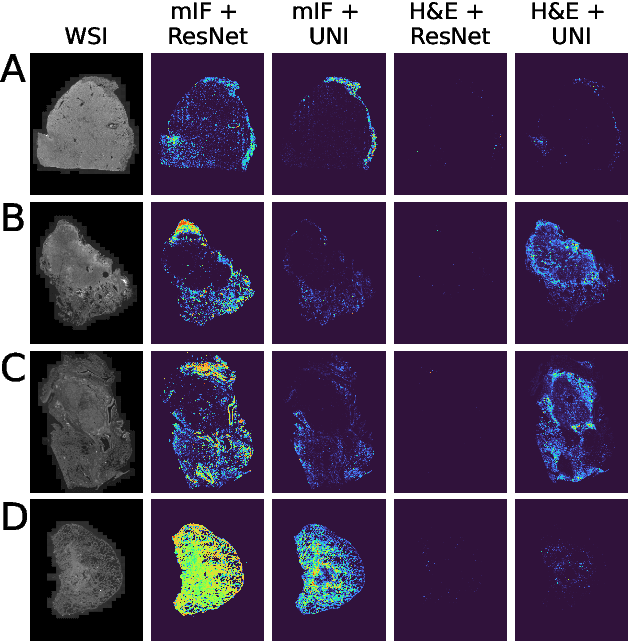
Though multiple instance learning (MIL) has been a foundational strategy in computational pathology for processing whole slide images (WSIs), current approaches are designed for traditional hematoxylin and eosin (H&E) slides rather than emerging multiplexed technologies. Here, we present an MIL strategy, the Fluoroformer module, that is specifically tailored to multiplexed WSIs by leveraging scaled dot-product attention (SDPA) to interpretably fuse information across disparate channels. On a cohort of 434 non-small cell lung cancer (NSCLC) samples, we show that the Fluoroformer both obtains strong prognostic performance and recapitulates immuno-oncological hallmarks of NSCLC. Our technique thereby provides a path for adapting state-of-the-art AI techniques to emerging spatial biology assays.
Combinatorial optimization of the coefficient of determination
Oct 12, 2024Robust correlation analysis is among the most critical challenges in statistics. Herein, we develop an efficient algorithm for selecting the $k$- subset of $n$ points in the plane with the highest coefficient of determination $\left( R^2 \right)$. Drawing from combinatorial geometry, we propose a method called the \textit{quadratic sweep} that consists of two steps: (i) projectively lifting the data points into $\mathbb R^5$ and then (ii) iterating over each linearly separable $k$-subset. Its basis is that the optimal set of outliers is separable from its complement in $\mathbb R^2$ by a conic section, which, in $\mathbb R^5$, can be found by a topological sweep in $\Theta \left( n^5 \log n \right)$ time. Although key proofs of quadratic separability remain underway, we develop strong mathematical intuitions for our conjectures, then experimentally demonstrate our method's optimality over several million trials up to $n=30$ without error. Implementations in Julia and fully seeded, reproducible experiments are available at https://github.com/marc-harary/QuadraticSweep.
Kirigami: large convolutional kernels improve deep learning-based RNA secondary structure prediction
Jun 04, 2024We introduce a novel fully convolutional neural network (FCN) architecture for predicting the secondary structure of ribonucleic acid (RNA) molecules. Interpreting RNA structures as weighted graphs, we employ deep learning to estimate the probability of base pairing between nucleotide residues. Unique to our model are its massive 11-pixel kernels, which we argue provide a distinct advantage for FCNs on the specialized domain of RNA secondary structures. On a widely adopted, standardized test set comprised of 1,305 molecules, the accuracy of our method exceeds that of current state-of-the-art (SOTA) secondary structure prediction software, achieving a Matthews Correlation Coefficient (MCC) over 11-40% higher than that of other leading methods on overall structures and 58-400% higher on pseudoknots specifically.