 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVariational phylogenetic inference with products over bipartitions
Feb 21, 2025Bayesian phylogenetics requires accurate and efficient approximation of posterior distributions over trees. In this work, we develop a variational Bayesian approach for ultrametric phylogenetic trees. We present a novel variational family based on coalescent times of a single-linkage clustering and derive a closed-form density of the resulting distribution over trees. Unlike existing methods for ultrametric trees, our method performs inference over all of tree space, it does not require any Markov chain Monte Carlo subroutines, and our variational family is differentiable. Through experiments on benchmark genomic datasets and an application to SARS-CoV-2, we demonstrate that our method achieves competitive accuracy while requiring significantly fewer gradient evaluations than existing state-of-the-art techniques.
Q-learning with online random forests
Apr 07, 2022
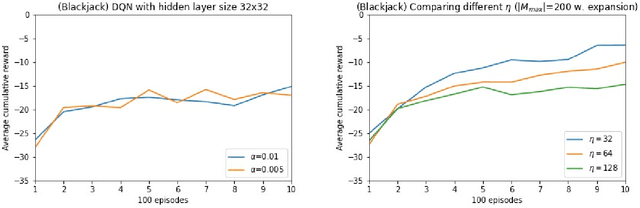

$Q$-learning is the most fundamental model-free reinforcement learning algorithm. Deployment of $Q$-learning requires approximation of the state-action value function (also known as the $Q$-function). In this work, we provide online random forests as $Q$-function approximators and propose a novel method wherein the random forest is grown as learning proceeds (through expanding forests). We demonstrate improved performance of our methods over state-of-the-art Deep $Q$-Networks in two OpenAI gyms (`blackjack' and `inverted pendulum') but not in the `lunar lander' gym. We suspect that the resilience to overfitting enjoyed by random forests recommends our method for common tasks that do not require a strong representation of the problem domain. We show that expanding forests (in which the number of trees increases as data comes in) improve performance, suggesting that expanding forests are viable for other applications of online random forests beyond the reinforcement learning setting.
Random Tessellation Forests
Jun 13, 2019



Space partitioning methods such as random forests and the Mondrian process are powerful machine learning methods for multi-dimensional and relational data, and are based on recursively cutting a domain. The flexibility of these methods is often limited by the requirement that the cuts be axis aligned. The Ostomachion process and the self-consistent binary space partitioning-tree process were recently introduced as generalizations of the Mondrian process for space partitioning with non-axis aligned cuts in the two dimensional plane. Motivated by the need for a multi-dimensional partitioning tree with non-axis aligned cuts, we propose the Random Tessellation Process (RTP), a framework that includes the Mondrian process and the binary space partitioning-tree process as special cases. We derive a sequential Monte Carlo algorithm for inference, and provide random forest methods. Our process is self-consistent and can relax axis-aligned constraints, allowing complex inter-dimensional dependence to be captured. We present a simulation study, and analyse gene expression data of brain tissue, showing improved accuracies over other methods.
A nonparametric HMM for genetic imputation and coalescent inference
Nov 02, 2016

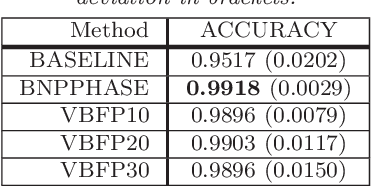

Genetic sequence data are well described by hidden Markov models (HMMs) in which latent states correspond to clusters of similar mutation patterns. Theory from statistical genetics suggests that these HMMs are nonhomogeneous (their transition probabilities vary along the chromosome) and have large support for self transitions. We develop a new nonparametric model of genetic sequence data, based on the hierarchical Dirichlet process, which supports these self transitions and nonhomogeneity. Our model provides a parameterization of the genetic process that is more parsimonious than other more general nonparametric models which have previously been applied to population genetics. We provide truncation-free MCMC inference for our model using a new auxiliary sampling scheme for Bayesian nonparametric HMMs. In a series of experiments on male X chromosome data from the Thousand Genomes Project and also on data simulated from a population bottleneck we show the benefits of our model over the popular finite model fastPHASE, which can itself be seen as a parametric truncation of our model. We find that the number of HMM states found by our model is correlated with the time to the most recent common ancestor in population bottlenecks. This work demonstrates the flexibility of Bayesian nonparametrics applied to large and complex genetic data.