 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNeural Network Based in Silico Simulation of Combustion Reactions
Nov 27, 2019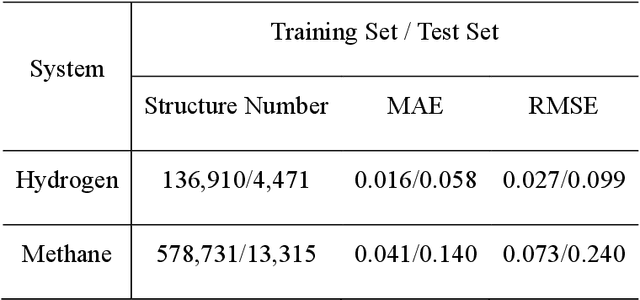
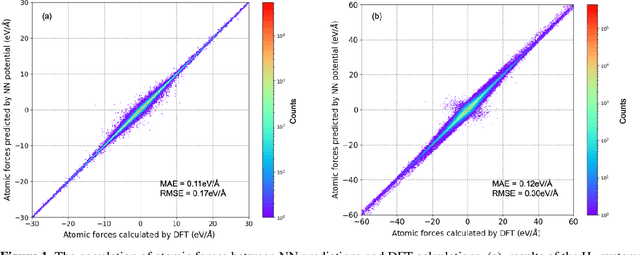
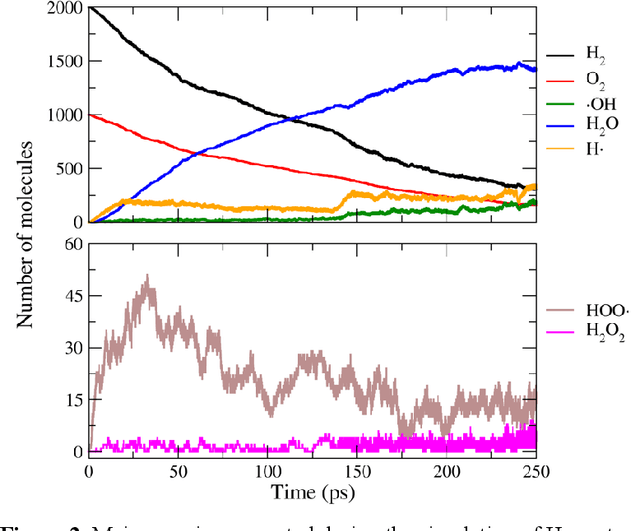
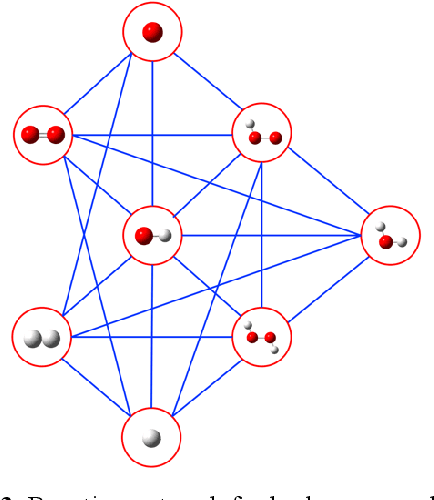
Understanding and prediction of the chemical reactions are fundamental demanding in the study of many complex chemical systems. Reactive molecular dynamics (MD) simulation has been widely used for this purpose as it can offer atomic details and can help us better interpret chemical reaction mechanisms. In this study, two reference datasets were constructed and corresponding neural network (NN) potentials were trained based on them. For given large-scale reaction systems, the NN potentials can predict the potential energy and atomic forces of DFT precision, while it is orders of magnitude faster than the conventional DFT calculation. With these two models, reactive MD simulations were performed to explore the combustion mechanisms of hydrogen and methane. Benefit from the high efficiency of the NN model, nanosecond MD trajectories for large-scale systems containing hundreds of atoms were produced and detailed combustion mechanism was obtained. Through further development, the algorithms in this study can be used to explore and discovery reaction mechanisms of many complex reaction systems, such as combustion, synthesis, and heterogeneous catalysis without any predefined reaction coordinates and elementary reaction steps.