 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConformation Clustering of Long MD Protein Dynamics with an Adversarial Autoencoder
May 31, 2018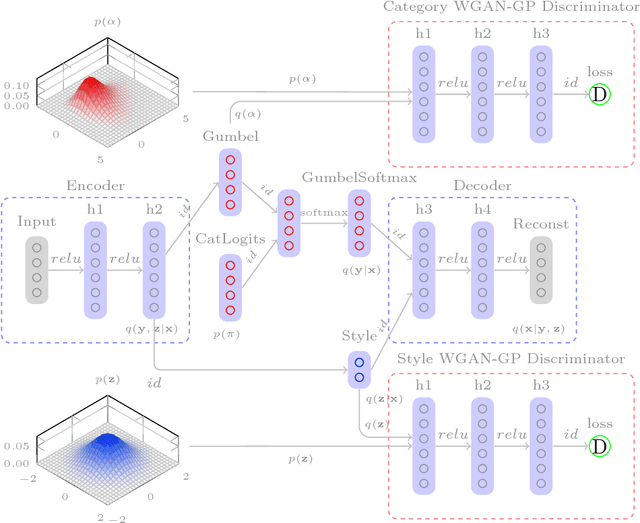



Recent developments in specialized computer hardware have greatly accelerated atomic level Molecular Dynamics (MD) simulations. A single GPU-attached cluster is capable of producing microsecond-length trajectories in reasonable amounts of time. Multiple protein states and a large number of microstates associated with folding and with the function of the protein can be observed as conformations sampled in the trajectories. Clustering those conformations, however, is needed for identifying protein states, evaluating transition rates and understanding protein behavior. In this paper, we propose a novel data-driven generative conformation clustering method based on the adversarial autoencoder (AAE) and provide the associated software implementation Cong. The method was tested using a 208 microseconds MD simulation of the fast-folding peptide Trp-Cage (20 residues) obtained from the D.E. Shaw Research Group. The proposed clustering algorithm identifies many of the salient features of the folding process by grouping a large number of conformations that share common features not easily identifiable in the trajectory.