 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLoop-Diffusion: an equivariant diffusion model for designing and scoring protein loops
Sep 26, 2024

Predicting protein functional characteristics from structure remains a central problem in protein science, with broad implications from understanding the mechanisms of disease to designing novel therapeutics. Unfortunately, current machine learning methods are limited by scarce and biased experimental data, and physics-based methods are either too slow to be useful, or too simplified to be accurate. In this work, we present Loop-Diffusion, an energy based diffusion model which leverages a dataset of general protein loops from the entire protein universe to learn an energy function that generalizes to functional prediction tasks. We evaluate Loop-Diffusion's performance on scoring TCR-pMHC interfaces and demonstrate state-of-the-art results in recognizing binding-enhancing mutations.
HERMES: Holographic Equivariant neuRal network model for Mutational Effect and Stability prediction
Jul 09, 2024
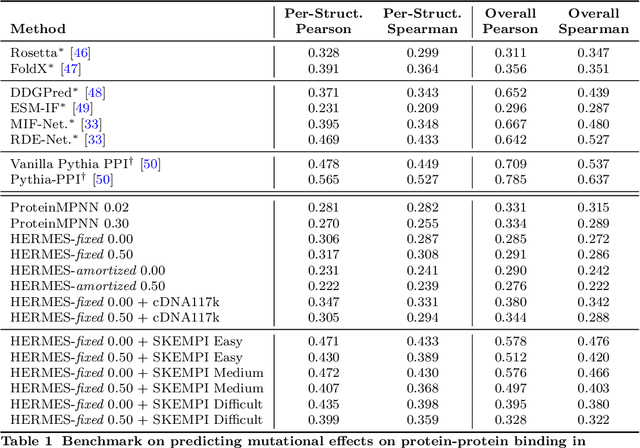


Predicting the stability and fitness effects of amino acid mutations in proteins is a cornerstone of biological discovery and engineering. Various experimental techniques have been developed to measure mutational effects, providing us with extensive datasets across a diverse range of proteins. By training on these data, traditional computational modeling and more recent machine learning approaches have advanced significantly in predicting mutational effects. Here, we introduce HERMES, a 3D rotationally equivariant structure-based neural network model for mutational effect and stability prediction. Pre-trained to predict amino acid propensity from its surrounding 3D structure, HERMES can be fine-tuned for mutational effects using our open-source code. We present a suite of HERMES models, pre-trained with different strategies, and fine-tuned to predict the stability effect of mutations. Benchmarking against other models shows that HERMES often outperforms or matches their performance in predicting mutational effect on stability, binding, and fitness. HERMES offers versatile tools for evaluating mutational effects and can be fine-tuned for specific predictive objectives.