 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSingle-neuron deep generative model uncovers underlying physics of neuronal activity in Ca imaging data
Jan 24, 2025
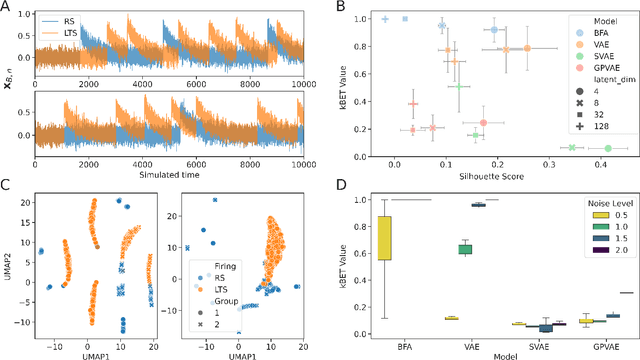

Calcium imaging has become a powerful alternative to electrophysiology for studying neuronal activity, offering spatial resolution and the ability to measure large populations of neurons in a minimally invasive manner. This technique has broad applications in neuroscience, neuroengineering, and medicine, enabling researchers to explore the relationship between neuron location and activity. Recent advancements in deep generative models (DGMs) have facilitated the modeling of neuronal population dynamics, uncovering latent representations that provide insights into behavior prediction and neuronal variance. However, these models often rely on spike inference algorithms and primarily focus on population-level dynamics, limiting their applicability for single-neuron analyses. To address this gap, we propose a novel framework for single-neuron representation learning using autoregressive variational autoencoders (AVAEs). Our approach embeds individual neurons' spatiotemporal signals into a reduced-dimensional space without the need for spike inference algorithms. The AVAE excels over traditional linear methods by generating more informative and discriminative latent representations, improving tasks such as visualization, clustering, and the understanding of neuronal activity. Additionally, the reconstruction performance of the AVAE outperforms the state of the art, demonstrating its ability to accurately recover the original fluorescence signal from the learned representation. Using realistic simulations, we show that our model captures underlying physical properties and connectivity patterns, enabling it to distinguish between different firing and connectivity types. These findings position the AVAE as a versatile and powerful tool for advancing single-neuron analysis and lays the groundwork for future integration of multimodal single-cell datasets in neuroscience.
Explainable AI model reveals disease-related mechanisms in single-cell RNA-seq data
Jan 07, 2025Neurodegenerative diseases (NDDs) are complex and lack effective treatment due to their poorly understood mechanism. The increasingly used data analysis from Single nucleus RNA Sequencing (snRNA-seq) allows to explore transcriptomic events at a single cell level, yet face challenges in interpreting the mechanisms underlying a disease. On the other hand, Neural Network (NN) models can handle complex data to offer insights but can be seen as black boxes with poor interpretability. In this context, explainable AI (XAI) emerges as a solution that could help to understand disease-associated mechanisms when combined with efficient NN models. However, limited research explores XAI in single-cell data. In this work, we implement a method for identifying disease-related genes and the mechanistic explanation of disease progression based on NN model combined with SHAP. We analyze available Huntington's disease (HD) data to identify both HD-altered genes and mechanisms by adding Gene Set Enrichment Analysis (GSEA) comparing two methods, differential gene expression analysis (DGE) and NN combined with SHAP approach. Our results show that DGE and SHAP approaches offer both common and differential sets of altered genes and pathways, reinforcing the usefulness of XAI methods for a broader perspective of disease.