 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCarbohydrate NMR chemical shift predictions using E(3) equivariant graph neural networks
Nov 21, 2023



Carbohydrates, vital components of biological systems, are well-known for their structural diversity. Nuclear Magnetic Resonance (NMR) spectroscopy plays a crucial role in understanding their intricate molecular arrangements and is essential in assessing and verifying the molecular structure of organic molecules. An important part of this process is to predict the NMR chemical shift from the molecular structure. This work introduces a novel approach that leverages E(3) equivariant graph neural networks to predict carbohydrate NMR spectra. Notably, our model achieves a substantial reduction in mean absolute error, up to threefold, compared to traditional models that rely solely on two-dimensional molecular structure. Even with limited data, the model excels, highlighting its robustness and generalization capabilities. The implications are far-reaching and go beyond an advanced understanding of carbohydrate structures and spectral interpretation. For example, it could accelerate research in pharmaceutical applications, biochemistry, and structural biology, offering a faster and more reliable analysis of molecular structures. Furthermore, our approach is a key step towards a new data-driven era in spectroscopy, potentially influencing spectroscopic techniques beyond NMR.
Machine Learning-Assisted Analysis of Small Angle X-ray Scattering
Nov 16, 2021
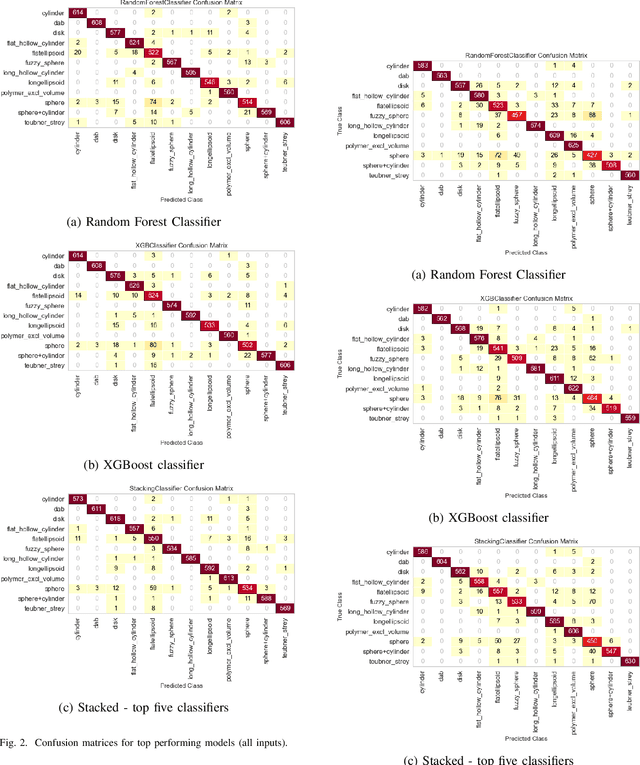
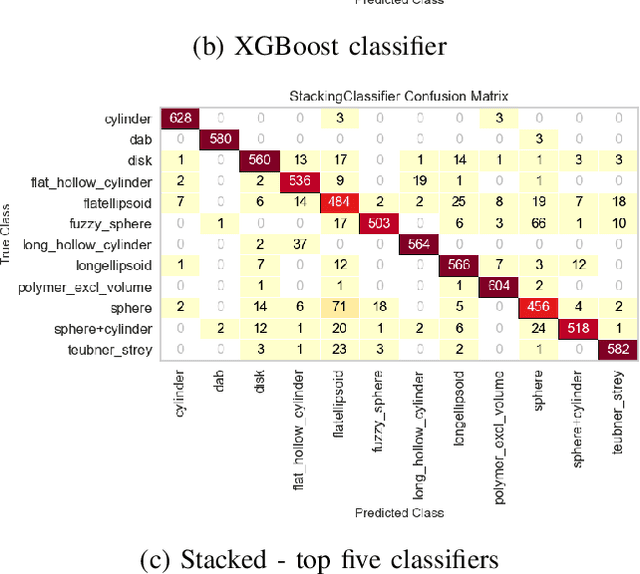
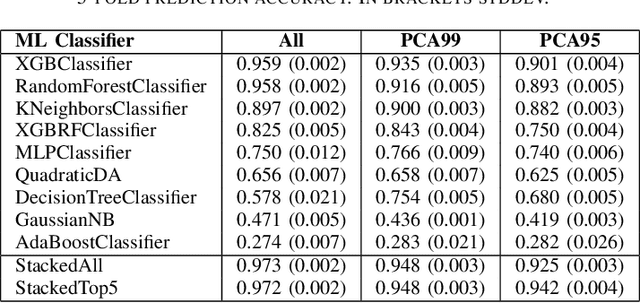
Small angle X-ray scattering (SAXS) is extensively used in materials science as a way of examining nanostructures. The analysis of experimental SAXS data involves mapping a rather simple data format to a vast amount of structural models. Despite various scientific computing tools to assist the model selection, the activity heavily relies on the SAXS analysts' experience, which is recognized as an efficiency bottleneck by the community. To cope with this decision-making problem, we develop and evaluate the open-source, Machine Learning-based tool SCAN (SCattering Ai aNalysis) to provide recommendations on model selection. SCAN exploits multiple machine learning algorithms and uses models and a simulation tool implemented in the SasView package for generating a well defined set of datasets. Our evaluation shows that SCAN delivers an overall accuracy of 95%-97%. The XGBoost Classifier has been identified as the most accurate method with a good balance between accuracy and training time. With eleven predefined structural models for common nanostructures and an easy draw-drop function to expand the number and types training models, SCAN can accelerate the SAXS data analysis workflow.