 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraphVAMPNet, using graph neural networks and variational approach to markov processes for dynamical modeling of biomolecules
Jan 12, 2022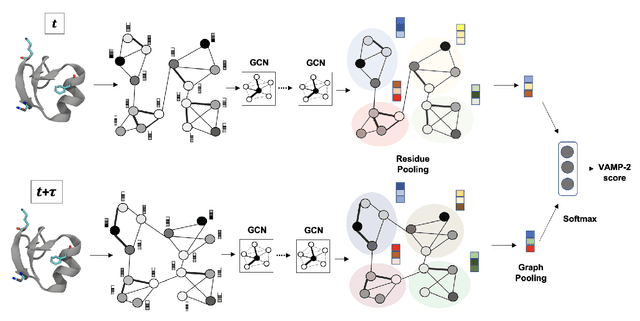

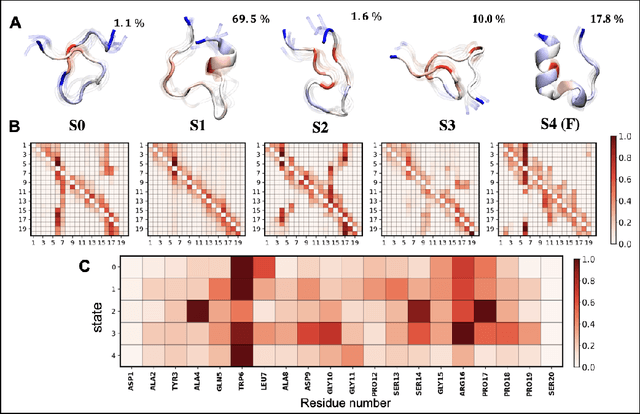
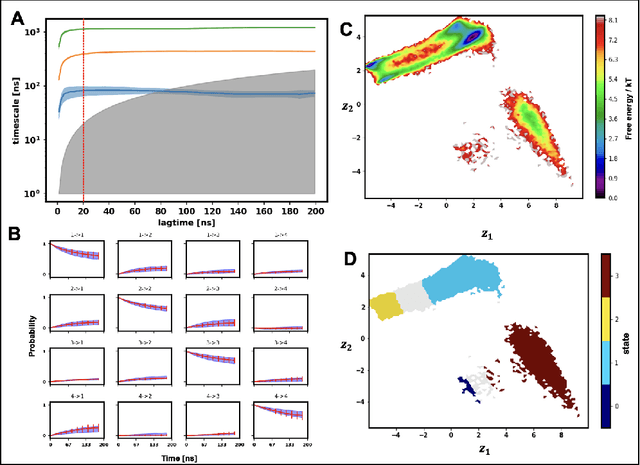
Finding low dimensional representation of data from long-timescale trajectories of biomolecular processes such as protein-folding or ligand-receptor binding is of fundamental importance and kinetic models such as Markov modeling have proven useful in describing the kinetics of these systems. Recently, an unsupervised machine learning technique called VAMPNet was introduced to learn the low dimensional representation and linear dynamical model in an end-to-end manner. VAMPNet is based on variational approach to Markov processes (VAMP) and relies on neural networks to learn the coarse-grained dynamics. In this contribution, we combine VAMPNet and graph neural networks to generate an end-to-end framework to efficiently learn high-level dynamics and metastable states from the long-timescale molecular dynamics trajectories. This method bears the advantages of graph representation learning and uses graph message passing operations to generate an embedding for each datapoint which is used in the VAMPNet to generate a coarse-grained representation. This type of molecular representation results in a higher resolution and more interpretable Markov model than the standard VAMPNet enabling a more detailed kinetic study of the biomolecular processes. Our GraphVAMPNet approach is also enhanced with an attention mechanism to find the important residues for classification into different metastable states.
Variational embedding of protein folding simulations using gaussian mixture variational autoencoders
Aug 27, 2021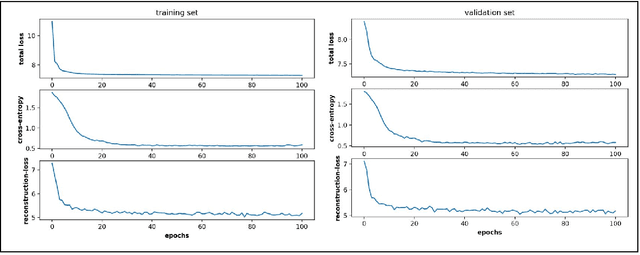
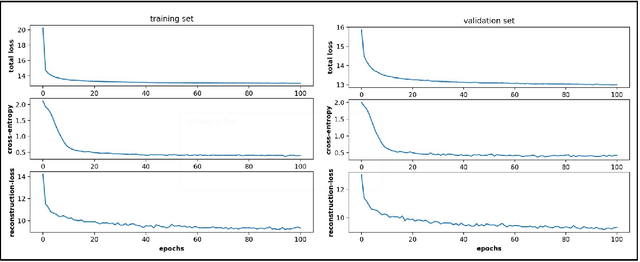


Conformational sampling of biomolecules using molecular dynamics simulations often produces large amount of high dimensional data that makes it difficult to interpret using conventional analysis techniques. Dimensionality reduction methods are thus required to extract useful and relevant information. Here we devise a machine learning method, Gaussian mixture variational autoencoder (GMVAE) that can simultaneously perform dimensionality reduction and clustering of biomolecular conformations in an unsupervised way. We show that GMVAE can learn a reduced representation of the free energy landscape of protein folding with highly separated clusters that correspond to the metastable states during folding. Since GMVAE uses a mixture of Gaussians as the prior, it can directly acknowledge the multi-basin nature of protein folding free-energy landscape. To make the model end-to-end differentialble, we use a Gumbel-softmax distribution. We test the model on three long-timescale protein folding trajectories and show that GMVAE embedding resembles the folding funnel with folded states down the funnel and unfolded states outer in the funnel path. Additionally, we show that the latent space of GMVAE can be used for kinetic analysis and Markov state models built on this embedding produce folding and unfolding timescales that are in close agreement with other rigorous dynamical embeddings such as time independent component analysis (TICA).