 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSEEN: Sharpening Explanations for Graph Neural Networks using Explanations from Neighborhoods
Jun 16, 2021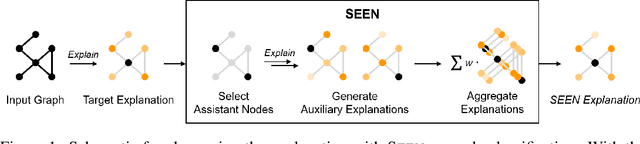



Explaining the foundations for predictions obtained from graph neural networks (GNNs) is critical for credible use of GNN models for real-world problems. Owing to the rapid growth of GNN applications, recent progress in explaining predictions from GNNs, such as sensitivity analysis, perturbation methods, and attribution methods, showed great opportunities and possibilities for explaining GNN predictions. In this study, we propose a method to improve the explanation quality of node classification tasks that can be applied in a post hoc manner through aggregation of auxiliary explanations from important neighboring nodes, named SEEN. Applying SEEN does not require modification of a graph and can be used with diverse explainability techniques due to its independent mechanism. Experiments on matching motif-participating nodes from a given graph show great improvement in explanation accuracy of up to 12.71% and demonstrate the correlation between the auxiliary explanations and the enhanced explanation accuracy through leveraging their contributions. SEEN provides a simple but effective method to enhance the explanation quality of GNN model outputs, and this method is applicable in combination with most explainability techniques.
InteractionNet: Modeling and Explaining of Noncovalent Protein-Ligand Interactions with Noncovalent Graph Neural Network and Layer-Wise Relevance Propagation
May 12, 2020



Expanding the scope of graph-based, deep-learning models to noncovalent protein-ligand interactions has earned increasing attention in structure-based drug design. Modeling the protein-ligand interactions with graph neural networks (GNNs) has experienced difficulties in the conversion of protein-ligand complex structures into the graph representation and left questions regarding whether the trained models properly learn the appropriate noncovalent interactions. Here, we proposed a GNN architecture, denoted as InteractionNet, which learns two separated molecular graphs, being covalent and noncovalent, through distinct convolution layers. We also analyzed the InteractionNet model with an explainability technique, i.e., layer-wise relevance propagation, for examination of the chemical relevance of the model's predictions. Separation of the covalent and noncovalent convolutional steps made it possible to evaluate the contribution of each step independently and analyze the graph-building strategy for noncovalent interactions. We applied InteractionNet to the prediction of protein-ligand binding affinity and showed that our model successfully predicted the noncovalent interactions in both performance and relevance in chemical interpretation.
Three-Dimensionally Embedded Graph Convolutional Network (3DGCN) for Molecule Interpretation
Dec 04, 2018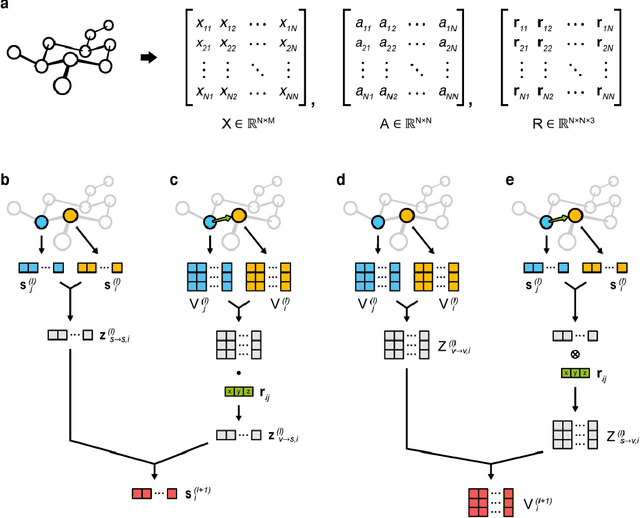
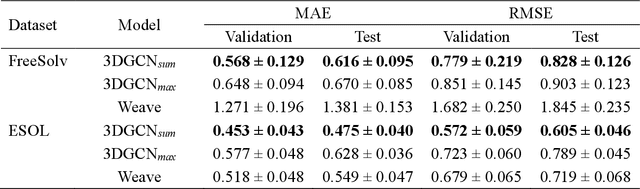
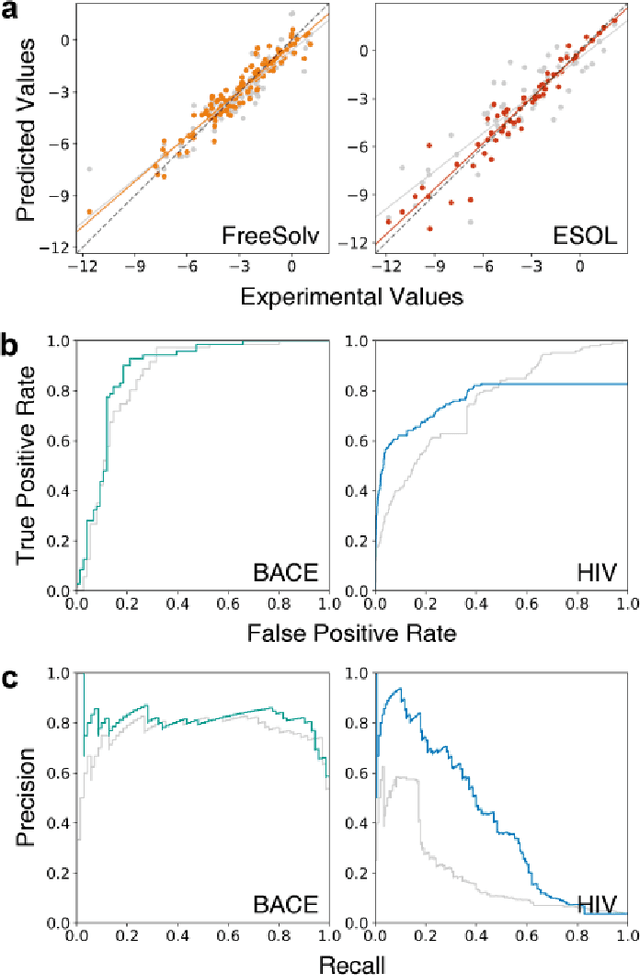
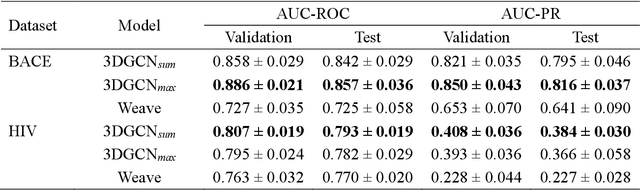
The target scope of graph convolutional networks (GCNs) for learning the graph representation of molecules has been expanded from chemical properties to biological activities, but the incorporation of the three-dimensional topology of molecules to the deep-learning models has not been explored. Most GCNs that achieve state-of-the-art performance rely only on the node distances, limiting the spatial information of molecules. In this work, we propose an advanced derivative of GCNs, coined a 3DGCN (three-dimensionally embedded graph convolutional network), which takes molecular graphs embedded in three-dimensional Euclidean space as inputs and recursively updates the scalar and vector features based on the relative positions of nodes. We demonstrate the learning capabilities of the 3DGCN using physical and biophysical prediction tasks.