 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFragmentRetro: A Quadratic Retrosynthetic Method Based on Fragmentation Algorithms
Sep 18, 2025Retrosynthesis, the process of deconstructing a target molecule into simpler precursors, is crucial for computer-aided synthesis planning (CASP). Widely adopted tree-search methods often suffer from exponential computational complexity. In this work, we introduce FragmentRetro, a novel retrosynthetic method that leverages fragmentation algorithms, specifically BRICS and r-BRICS, combined with stock-aware exploration and pattern fingerprint screening to achieve quadratic complexity. FragmentRetro recursively combines molecular fragments and verifies their presence in a building block set, providing sets of fragment combinations as retrosynthetic solutions. We present the first formal computational analysis of retrosynthetic methods, showing that tree search exhibits exponential complexity $O(b^h)$, DirectMultiStep scales as $O(h^6)$, and FragmentRetro achieves $O(h^2)$, where $h$ represents the number of heavy atoms in the target molecule and $b$ is the branching factor for tree search. Evaluations on PaRoutes, USPTO-190, and natural products demonstrate that FragmentRetro achieves high solved rates with competitive runtime, including cases where tree search fails. The method benefits from fingerprint screening, which significantly reduces substructure matching complexity. While FragmentRetro focuses on efficiently identifying fragment-based solutions rather than full reaction pathways, its computational advantages and ability to generate strategic starting candidates establish it as a powerful foundational component for scalable and automated synthesis planning.
A Model-Centric Review of Deep Learning for Protein Design
Feb 26, 2025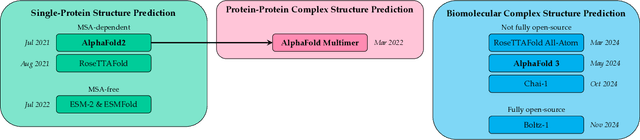
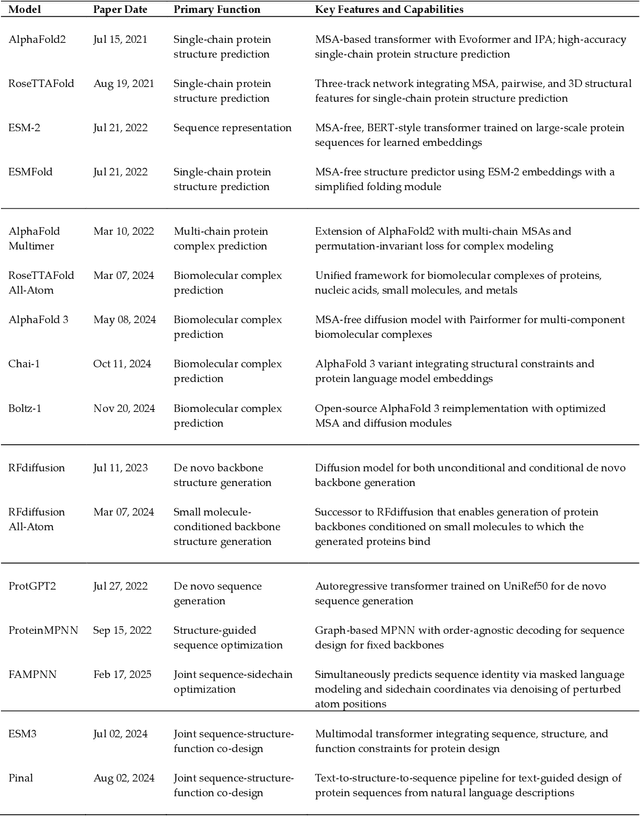
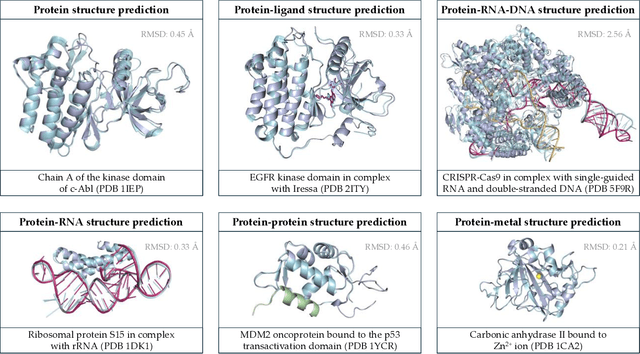
Deep learning has transformed protein design, enabling accurate structure prediction, sequence optimization, and de novo protein generation. Advances in single-chain protein structure prediction via AlphaFold2, RoseTTAFold, ESMFold, and others have achieved near-experimental accuracy, inspiring successive work extended to biomolecular complexes via AlphaFold Multimer, RoseTTAFold All-Atom, AlphaFold 3, Chai-1, Boltz-1 and others. Generative models such as ProtGPT2, ProteinMPNN, and RFdiffusion have enabled sequence and backbone design beyond natural evolution-based limitations. More recently, joint sequence-structure co-design models, including ESM3, have integrated both modalities into a unified framework, resulting in improved designability. Despite these advances, challenges still exist pertaining to modeling sequence-structure-function relationships and ensuring robust generalization beyond the regions of protein space spanned by the training data. Future advances will likely focus on joint sequence-structure-function co-design frameworks that are able to model the fitness landscape more effectively than models that treat these modalities independently. Current capabilities, coupled with the dizzying rate of progress, suggest that the field will soon enable rapid, rational design of proteins with tailored structures and functions that transcend the limitations imposed by natural evolution. In this review, we discuss the current capabilities of deep learning methods for protein design, focusing on some of the most revolutionary and capable models with respect to their functionality and the applications that they enable, leading up to the current challenges of the field and the optimal path forward.
CardioGenAI: A Machine Learning-Based Framework for Re-Engineering Drugs for Reduced hERG Liability
Mar 12, 2024Drug-induced cardiotoxicity is a major health concern which can lead to serious adverse effects including life-threatening cardiac arrhythmias via the blockade of the voltage-gated hERG potassium ion channel. It is therefore of tremendous interest to develop advanced methods to identify hERG-active compounds in early stages of drug development, as well as to optimize commercially available drugs for reduced hERG activity. In this work, we present CardioGenAI, a machine learning-based framework for re-engineering both developmental and marketed drugs for reduced hERG activity while preserving their pharmacological activity. The framework incorporates novel state-of-the-art discriminative models for predicting hERG channel activity, as well as activity against the voltage-gated NaV1.5 and CaV1.2 channels due to their potential implications in modulating the arrhythmogenic potential induced by hERG channel blockade. These models can also serve independently as effective components of a virtual screening pipeline. We applied the complete framework to pimozide, an FDA-approved antipsychotic agent that demonstrates high affinity to the hERG channel, and generated 100 refined candidates. Remarkably, among the candidates is fluspirilene, a compound which is of the same class of drugs (diphenylmethanes) as pimozide and therefore has similar pharmacological activity, yet exhibits over 700-fold weaker binding to hERG. We have made all of our software open-source to facilitate integration of the CardioGenAI framework for molecular hypothesis generation into drug discovery workflows.
ChemSpaceAL: An Efficient Active Learning Methodology Applied to Protein-Specific Molecular Generation
Sep 11, 2023The incredible capabilities of generative artificial intelligence models have inevitably led to their application in the domain of drug discovery. It is therefore of tremendous interest to develop methodologies that enhance the abilities and applicability of these powerful tools. In this work, we present a novel and efficient semi-supervised active learning methodology that allows for the fine-tuning of a generative model with respect to an objective function by strategically operating within a constructed representation of the sample space. In the context of targeted molecular generation, we demonstrate the ability to fine-tune a GPT-based molecular generator with respect to an attractive interaction-based scoring function by strategically operating within a chemical space proxy, thereby maximizing attractive interactions between the generated molecules and a protein target. Importantly, our approach does not require the individual evaluation of all data points that are used for fine-tuning, enabling the incorporation of computationally expensive metrics. We are hopeful that the inherent generality of this methodology ensures that it will remain applicable as this exciting field evolves. To facilitate implementation and reproducibility, we have made all of our software available through the open-source ChemSpaceAL Python package.
Quantum Convolutional Neural Networks for Multi-Channel Supervised Learning
May 30, 2023As the rapidly evolving field of machine learning continues to produce incredibly useful tools and models, the potential for quantum computing to provide speed up for machine learning algorithms is becoming increasingly desirable. In particular, quantum circuits in place of classical convolutional filters for image detection-based tasks are being investigated for the ability to exploit quantum advantage. However, these attempts, referred to as quantum convolutional neural networks (QCNNs), lack the ability to efficiently process data with multiple channels and therefore are limited to relatively simple inputs. In this work, we present a variety of hardware-adaptable quantum circuit ansatzes for use as convolutional kernels, and demonstrate that the quantum neural networks we report outperform existing QCNNs on classification tasks involving multi-channel data. We envision that the ability of these implementations to effectively learn inter-channel information will allow quantum machine learning methods to operate with more complex data. This work is available as open source at https://github.com/anthonysmaldone/QCNN-Multi-Channel-Supervised-Learning.
HAC-Net: A Hybrid Attention-Based Convolutional Neural Network for Highly Accurate Protein-Ligand Binding Affinity Prediction
Dec 23, 2022



Applying deep learning concepts from image detection and graph theory has greatly advanced protein-ligand binding affinity prediction, a challenge with enormous ramifications for both drug discovery and protein engineering. We build upon these advances by designing a novel deep learning architecture consisting of a 3-dimensional convolutional neural network utilizing channel-wise attention and two graph convolutional networks utilizing attention-based aggregation of node features. HAC-Net (Hybrid Attention-Based Convolutional Neural Network) obtains state-of-the-art results on the PDBbind v.2016 core set, the most widely recognized benchmark in the field. We extensively assess the generalizability of our model using multiple train-test splits, each of which maximizes differences between either protein structures, protein sequences, or ligand extended-connectivity fingerprints. Furthermore, we perform 10-fold cross-validation with a similarity cutoff between SMILES strings of ligands in the training and test sets, and also evaluate the performance of HAC-Net on lower-quality data. We envision that this model can be extended to a broad range of supervised learning problems related to structure-based biomolecular property prediction. All of our software is available as open source at https://github.com/gregory-kyro/HAC-Net/.