 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRapid Exploration of a 32.5M Compound Chemical Space with Active Learning to Discover Density Functional Approximation Insensitive and Synthetically Accessible Transitional Metal Chromophores
Aug 10, 2022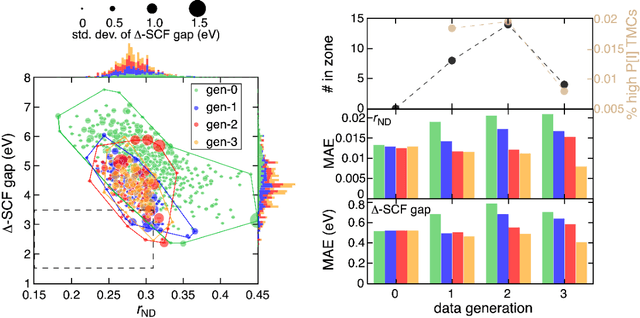
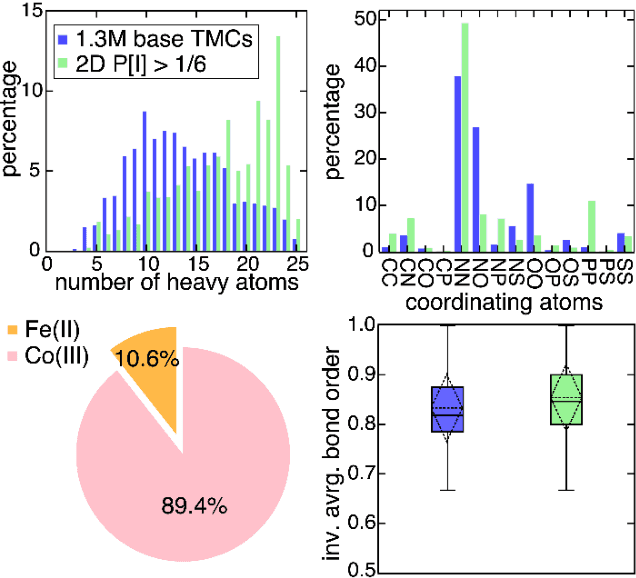
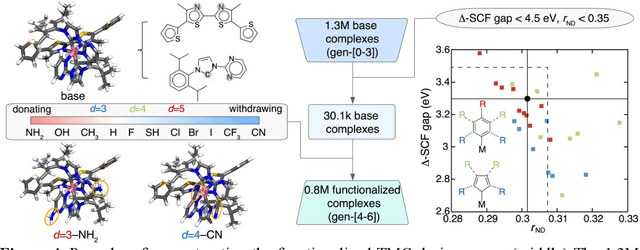
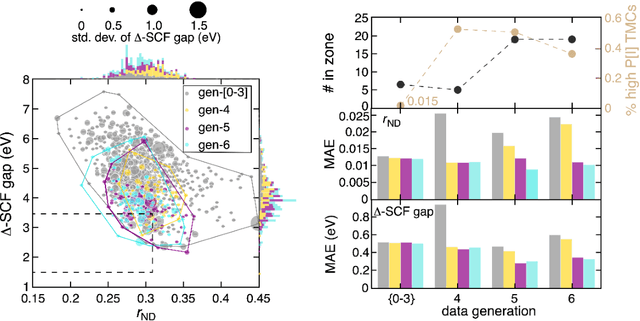
Two outstanding challenges for machine learning (ML) accelerated chemical discovery are the synthesizability of candidate molecules or materials and the fidelity of the data used in ML model training. To address the first challenge, we construct a hypothetical design space of 32.5M transition metal complexes (TMCs), in which all of the constituent fragments (i.e., metals and ligands) and ligand symmetries are synthetically accessible. To address the second challenge, we search for consensus in predictions among 23 density functional approximations across multiple rungs of Jacob's ladder. To accelerate the screening of these 32.5M TMCs, we use efficient global optimization to sample candidate low-spin chromophores that simultaneously have low absorption energies and low static correlation. Despite the scarcity (i.e., $<$ 0.01\%) of potential chromophores in this large chemical space, we identify transition metal chromophores with high likelihood (i.e., $>$ 10\%) as the ML models improve during active learning. This represents a 1,000 fold acceleration in discovery corresponding to discoveries in days instead of years. Analyses of candidate chromophores reveal a preference for Co(III) and large, strong-field ligands with more bond saturation. We compute the absorption spectra of promising chromophores on the Pareto front by time-dependent density functional theory calculations and verify that two thirds of them have desired excited state properties. Although these complexes have never been experimentally explored, their constituent ligands demonstrated interesting optical properties in literature, exemplifying the effectiveness of our construction of realistic TMC design space and active learning approach.