 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStructure-based drug design with geometric deep learning
Oct 19, 2022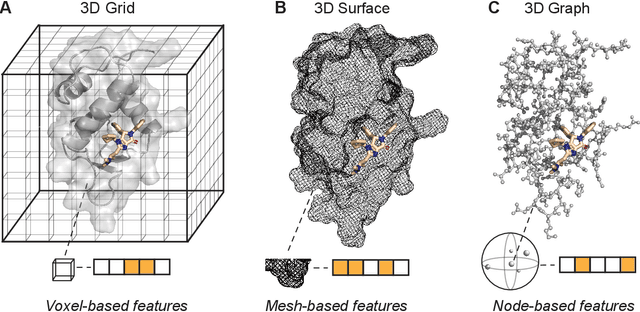
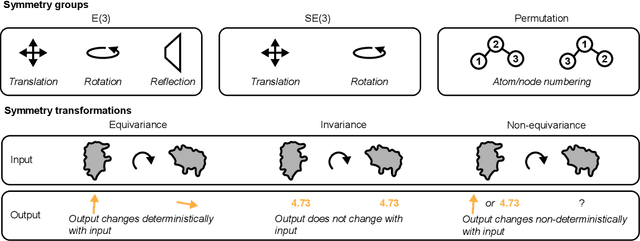

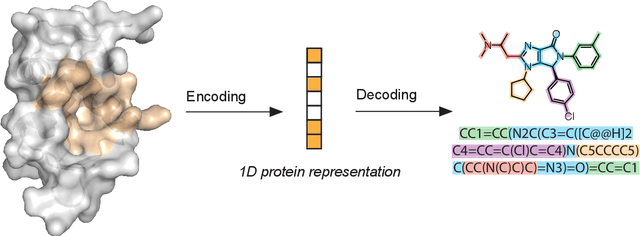
Structure-based drug design uses three-dimensional geometric information of macromolecules, such as proteins or nucleic acids, to identify suitable ligands. Geometric deep learning, an emerging concept of neural-network-based machine learning, has been applied to macromolecular structures. This review provides an overview of the recent applications of geometric deep learning in bioorganic and medicinal chemistry, highlighting its potential for structure-based drug discovery and design. Emphasis is placed on molecular property prediction, ligand binding site and pose prediction, and structure-based de novo molecular design. The current challenges and opportunities are highlighted, and a forecast of the future of geometric deep learning for drug discovery is presented.