 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA hybrid quantum-classical fusion neural network to improve protein-ligand binding affinity predictions for drug discovery
Sep 06, 2023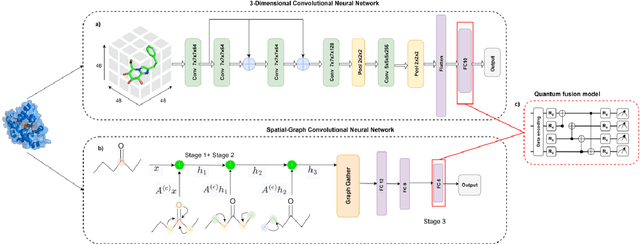
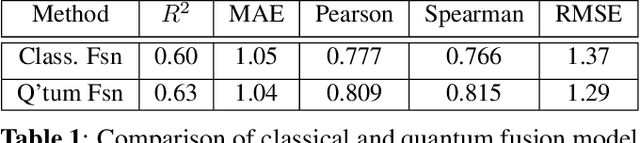
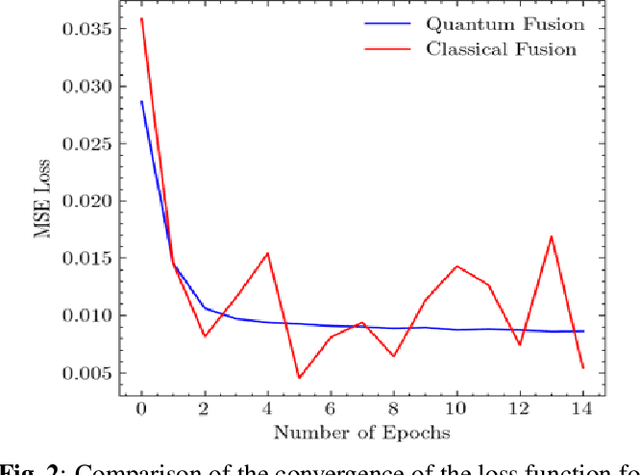
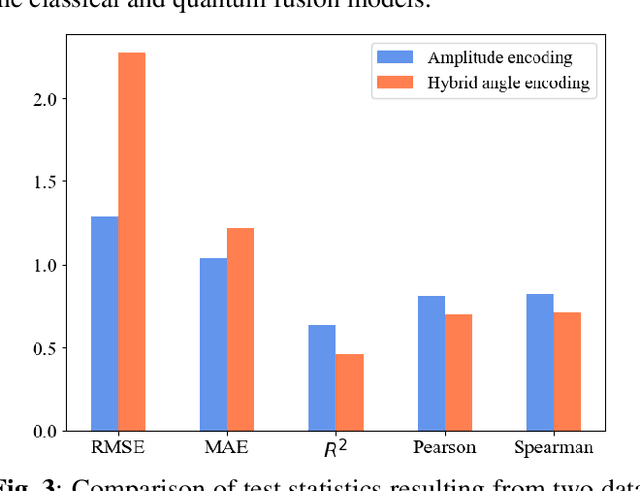
The field of drug discovery hinges on the accurate prediction of binding affinity between prospective drug molecules and target proteins, especially when such proteins directly influence disease progression. However, estimating binding affinity demands significant financial and computational resources. While state-of-the-art methodologies employ classical machine learning (ML) techniques, emerging hybrid quantum machine learning (QML) models have shown promise for enhanced performance, owing to their inherent parallelism and capacity to manage exponential increases in data dimensionality. Despite these advances, existing models encounter issues related to convergence stability and prediction accuracy. This paper introduces a novel hybrid quantum-classical deep learning model tailored for binding affinity prediction in drug discovery. Specifically, the proposed model synergistically integrates 3D and spatial graph convolutional neural networks within an optimized quantum architecture. Simulation results demonstrate a 6% improvement in prediction accuracy relative to existing classical models, as well as a significantly more stable convergence performance compared to previous classical approaches.
Hybrid quantum-classical convolutional neural networks to improve molecular protein binding affinity predictions
Jan 18, 2023



One of the main challenges in drug discovery is to find molecules that bind specifically and strongly to their target protein while having minimal binding to other proteins. By predicting binding affinity, it is possible to identify the most promising candidates from a large pool of potential compounds, reducing the number of compounds that need to be tested experimentally. Recently, deep learning methods have shown superior performance than traditional computational methods for making accurate predictions on large datasets. However, the complexity and time-consuming nature of these methods have limited their usage and development. Quantum machine learning is an emerging technology that has the potential to improve many classical machine learning algorithms. In this work we present a hybrid quantum-classical convolutional neural network, which is able to reduce by 20% the complexity of the classical network while maintaining optimal performance in the predictions. Additionally, it results in a significant time savings of up to 40% in the training process, which means a meaningful speed up of the drug discovery process.