 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomated, predictive, and interpretable inference of C. elegans escape dynamics
Sep 25, 2018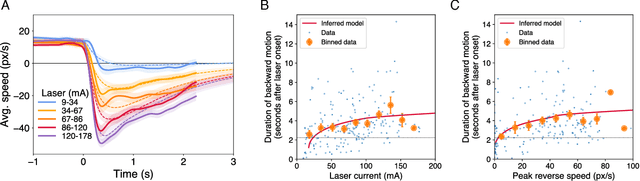
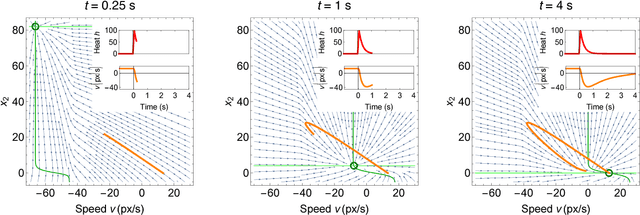
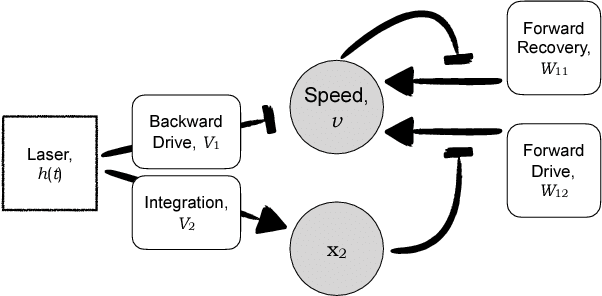
The roundworm C. elegans exhibits robust escape behavior in response to rapidly rising temperature. The behavior lasts for a few seconds, shows history dependence, involves both sensory and motor systems, and is too complicated to model mechanistically using currently available knowledge. Instead we model the process phenomenologically, and we use the Sir Isaac dynamical inference platform to infer the model in a fully automated fashion directly from experimental data. The inferred model requires incorporation of an unobserved dynamical variable, and is biologically interpretable. The model makes accurate predictions about the dynamics of the worm behavior, and it can be used to characterize the functional logic of the dynamical system underlying the escape response. This work illustrates the power of modern artificial intelligence to aid in discovery of accurate and interpretable models of complex natural systems.
Automated adaptive inference of coarse-grained dynamical models in systems biology
Apr 24, 2014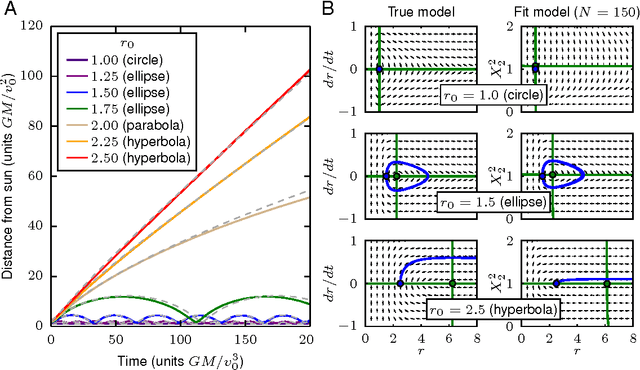
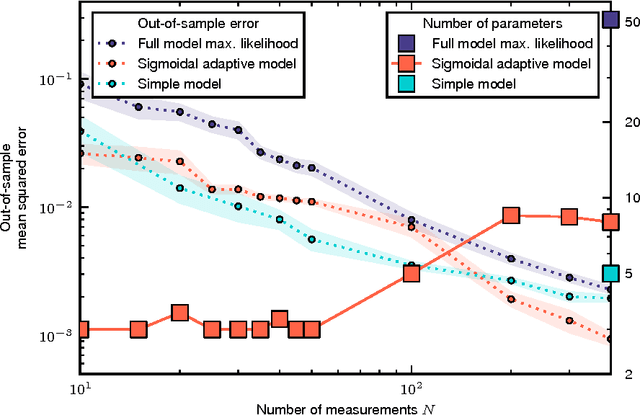
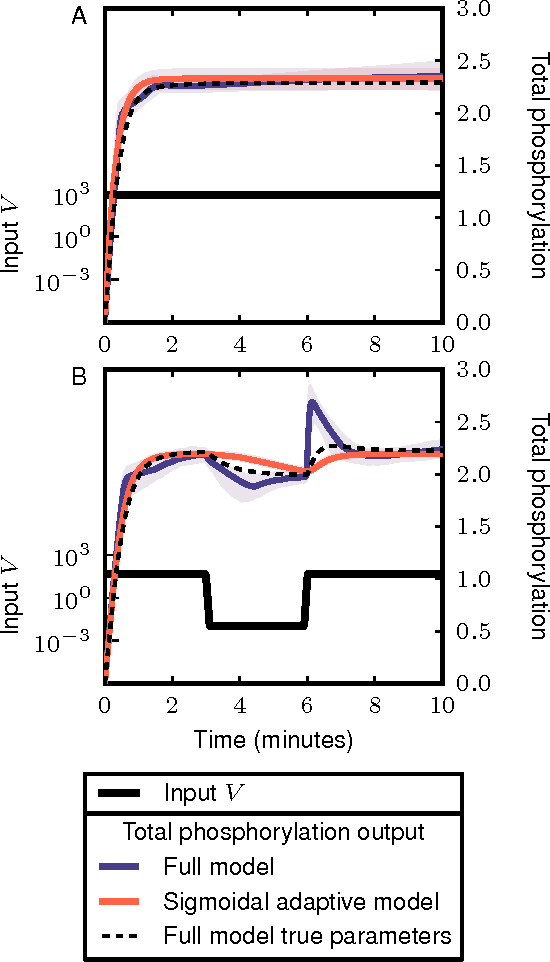
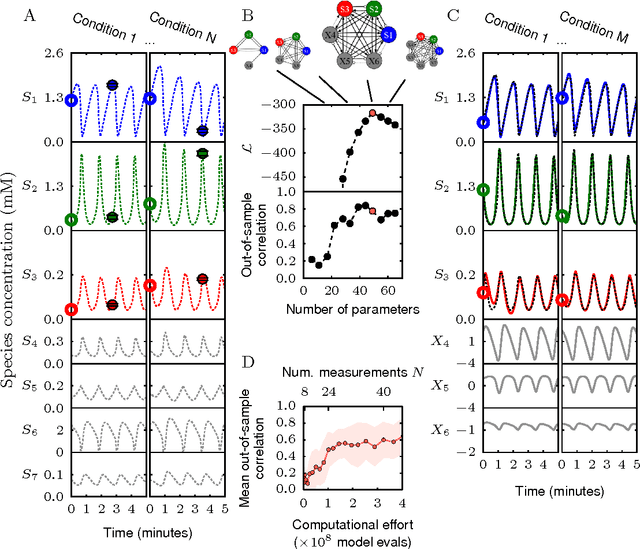
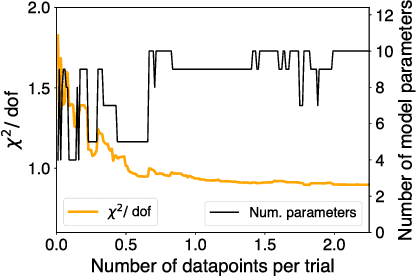
Cellular regulatory dynamics is driven by large and intricate networks of interactions at the molecular scale, whose sheer size obfuscates understanding. In light of limited experimental data, many parameters of such dynamics are unknown, and thus models built on the detailed, mechanistic viewpoint overfit and are not predictive. At the other extreme, simple ad hoc models of complex processes often miss defining features of the underlying systems. Here we propose an approach that instead constructs phenomenological, coarse-grained models of network dynamics that automatically adapt their complexity to the amount of available data. Such adaptive models lead to accurate predictions even when microscopic details of the studied systems are unknown due to insufficient data. The approach is computationally tractable, even for a relatively large number of dynamical variables, allowing its software realization, named Sir Isaac, to make successful predictions even when important dynamic variables are unobserved. For example, it matches the known phase space structure for simulated planetary motion data, avoids overfitting in a complex biological signaling system, and produces accurate predictions for a yeast glycolysis model with only tens of data points and over half of the interacting species unobserved.