 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBand gap prediction for large organic crystal structures with machine learning
Oct 30, 2018

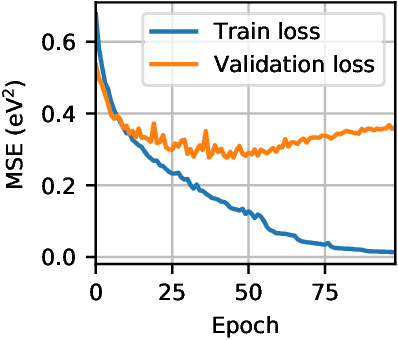

Machine learning models are capable of capturing the structure-property relationship from a dataset of computationally demanding ab initio calculations. In fact, machine learning models have reached chemical accuracy on small organic molecules contained in the popular QM9 dataset. At the same time, the domain of large crystal structures remains rather unexplored. Over the past two years, the Organic Materials Database (OMDB) has hosted a growing number of electronic properties of previously synthesized organic crystal structures. The complexity of the organic crystals contained within the OMDB, which have on average 85 atoms per unit cell, makes this database a challenging platform for machine learning applications. In this paper, we focus on predicting the band gap which represents one of the basic properties of a crystalline material. With this aim, we release a consistent dataset of 12500 crystal structures and their corresponding DFT band gap freely available for download at https://omdb.diracmaterials.org/dataset. We run two recent machine learning models, kernel ridge regression with the Smooth Overlap of Atomic Positions (SOAP) kernel and the deep learning model SchNet, on this new dataset and find that an ensemble of these two models reaches mean absolute error (MAE) of 0.361 eV, which corresponds to a percentage error of 12% on the average band gap of 3.03 eV. The models also provide chemical insights into the data. For example, by visualizing the SOAP kernel similarity between the crystals, different clusters of materials can be identified, such as organic metals or semiconductors. Finally, the trained models are employed to predict the band gap for 260092 materials contained within the Crystallography Open Database (COD) and made available online so the predictions can be obtained for any arbitrary crystal structure uploaded by a user.