 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEncoding Domain Knowledge in Multi-view Latent Variable Models: A Bayesian Approach with Structured Sparsity
Apr 13, 2022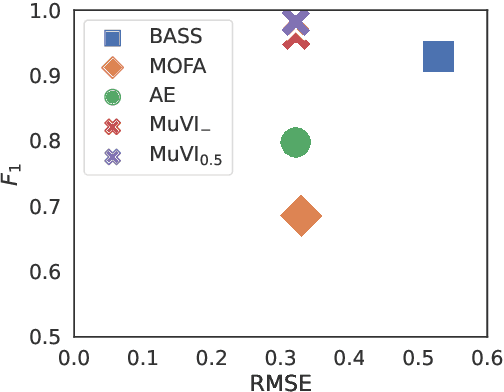
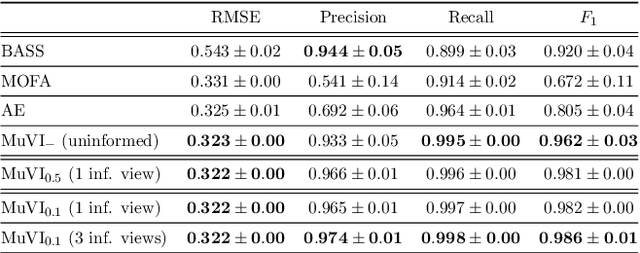
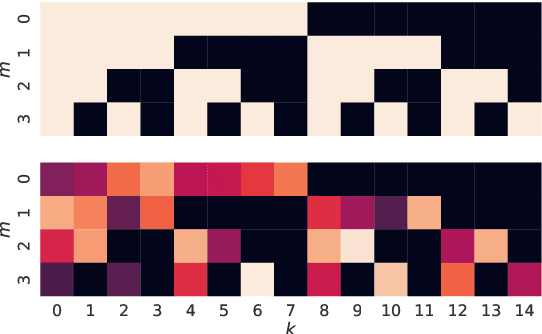

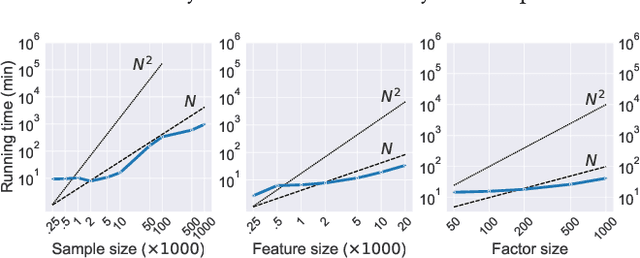
Many real-world systems are described not only by data from a single source but via multiple data views. For example, in genomic medicine, a patient can be described by data from different molecular layers. This raises the need for multi-view models that are able to disentangle variation within and across data views in an interpretable manner. Latent variable models with structured sparsity are a commonly used tool to address this modeling task but interpretability is cumbersome since it requires a direct inspection and interpretation of each factor via a specialized domain expert. Here, we propose MuVI, a novel approach for domain-informed multi-view latent variable models, facilitating the analysis of multi-view data in an inherently explainable manner. We demonstrate that our model (i) is able to integrate noisy domain expertise in form of feature sets, (ii) is robust to noise in the encoded domain knowledge, (iii) results in identifiable factors and (iv) is able to infer interpretable and biologically meaningful axes of variation in a real-world multi-view dataset of cancer patients.
Encoding Domain Information with Sparse Priors for Inferring Explainable Latent Variables
Jul 08, 2021


Latent variable models are powerful statistical tools that can uncover relevant variation between patients or cells, by inferring unobserved hidden states from observable high-dimensional data. A major shortcoming of current methods, however, is their inability to learn sparse and interpretable hidden states. Additionally, in settings where partial knowledge on the latent structure of the data is readily available, a statistically sound integration of prior information into current methods is challenging. To address these issues, we propose spex-LVM, a factorial latent variable model with sparse priors to encourage the inference of explainable factors driven by domain-relevant information. spex-LVM utilizes existing knowledge of curated biomedical pathways to automatically assign annotated attributes to latent factors, yielding interpretable results tailored to the corresponding domain of interest. Evaluations on simulated and real single-cell RNA-seq datasets demonstrate that our model robustly identifies relevant structure in an inherently explainable manner, distinguishes technical noise from sources of biomedical variation, and provides dataset-specific adaptations of existing pathway annotations. Implementation is available at https://github.com/MLO-lab/spexlvm.