 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNonlinear Discovery of Slow Molecular Modes using Hierarchical Dynamics Encoders
Feb 09, 2019

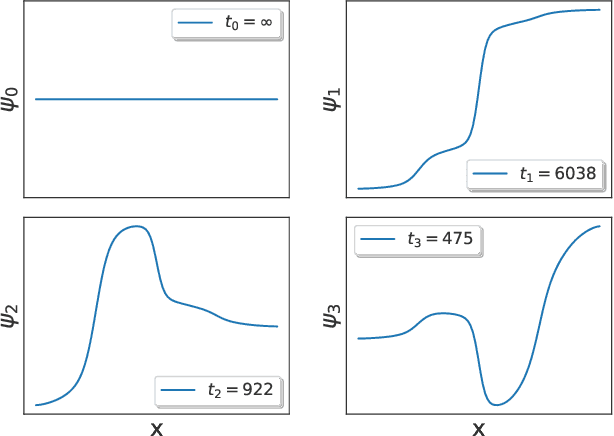
The success of enhanced sampling molecular simulations that accelerate along collective variables (CVs) is predicated on the availability of variables coincident with the slow collective motions governing the long-time conformational dynamics of a system. It is challenging to intuit these slow CVs for all but the simplest molecular systems, and their data-driven discovery directly from molecular simulation trajectories has been a central focus of the molecular simulation community to both unveil the important physical mechanisms and to drive enhanced sampling. In this work, we introduce hierarchical dynamics encoder (HDE) as a deep learning architecture that learns nonlinear CV approximants to the leading slow eigenfunctions of the spectral decomposition of the transfer operator that evolves equilibrium-scaled probability distributions through time. Orthogonality of the learned CVs is naturally imposed within network training without added regularization. The CVs are inherently explicit and differentiable functions of the input coordinates making them well-suited to use in enhanced sampling calculations. We demonstrate the utility of HDEs in capturing parsimonious nonlinear representations of complex system dynamics in applications to 1D and 2D toy systems where the true eigenfunctions are exactly calculable and to molecular dynamics simulations of alanine dipeptide and the WW domain protein.
Molecular enhanced sampling with autoencoders: On-the-fly collective variable discovery and accelerated free energy landscape exploration
Oct 30, 2018


Macromolecular and biomolecular folding landscapes typically contain high free energy barriers that impede efficient sampling of configurational space by standard molecular dynamics simulation. Biased sampling can artificially drive the simulation along pre-specified collective variables (CVs), but success depends critically on the availability of good CVs associated with the important collective dynamical motions. Nonlinear machine learning techniques can identify such CVs but typically do not furnish an explicit relationship with the atomic coordinates necessary to perform biased sampling. In this work, we employ auto-associative artificial neural networks ("autoencoders") to learn nonlinear CVs that are explicit and differentiable functions of the atomic coordinates. Our approach offers substantial speedups in exploration of configurational space, and is distinguished from exiting approaches by its capacity to simultaneously discover and directly accelerate along data-driven CVs. We demonstrate the approach in simulations of alanine dipeptide and Trp-cage, and have developed an open-source and freely-available implementation within OpenMM.