 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerated reactive transport simulations in heterogeneous porous medium using Reaktoro and Firedrake
Aug 17, 2020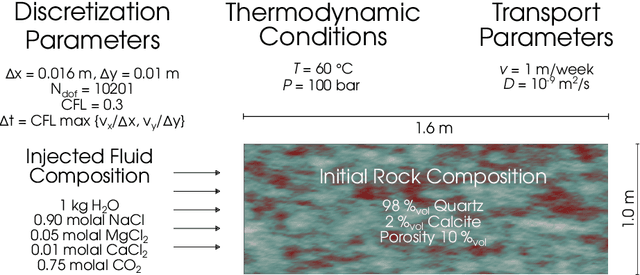

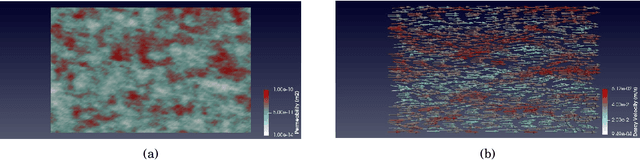

Geochemical reaction calculations in reactive transport modeling are costly in general. They become more expensive the more complex is the chemical system and the activity models used to describe the non-ideal thermodynamic behavior of its phases. Accounting for many aqueous species, gases, and minerals also contributes to more expensive computations. This work investigates the performance of the on-demand machine learning (ODML) algorithm presented in Allan etal. (2020) when applied to different reactive transport problems in heterogeneous porous media. We demonstrate that the ODML algorithm enables faster chemical equilibrium calculations by one to three orders of magnitude. This, in turn, significantly accelerates the entire reactive transport simulations. The numerical experiments are carried out using the coupling of two open-source software packages: Firedrake (Rathgeber, 2016) and Reaktoro (Leal, 2015).
Ultra-Fast Reactive Transport Simulations When Chemical Reactions Meet Machine Learning: Chemical Equilibrium
Aug 16, 2017
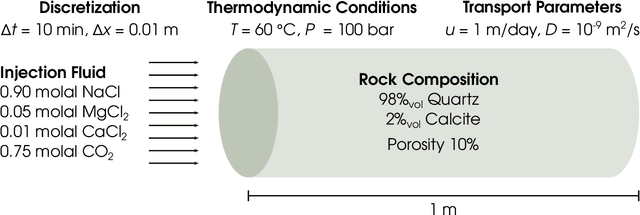
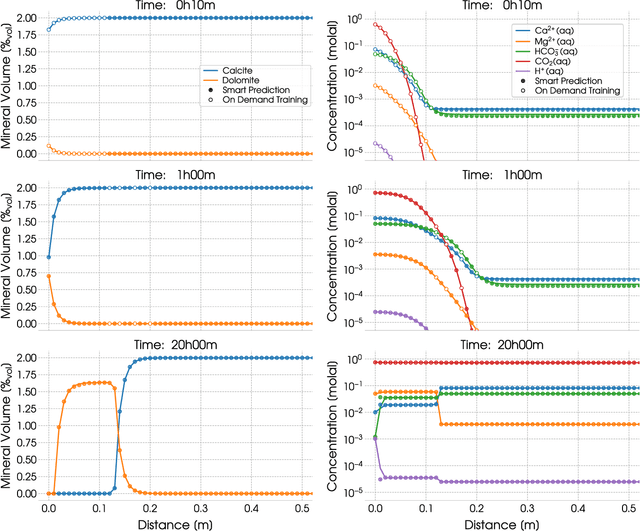
During reactive transport modeling, the computational cost associated with chemical reaction calculations is often 10-100 times higher than that of transport calculations. Most of these costs results from chemical equilibrium calculations that are performed at least once in every mesh cell and at every time step of the simulation. Calculating chemical equilibrium is an iterative process, where each iteration is in general so computationally expensive that even if every calculation converged in a single iteration, the resulting speedup would not be significant. Thus, rather than proposing a fast-converging numerical method for solving chemical equilibrium equations, we present a machine learning method that enables new equilibrium states to be quickly and accurately estimated, whenever a previous equilibrium calculation with similar input conditions has been performed. We demonstrate the use of this smart chemical equilibrium method in a reactive transport modeling example and show that, even at early simulation times, the majority of all equilibrium calculations are quickly predicted and, after some time steps, the machine-learning-accelerated chemical solver has been fully trained to rapidly perform all subsequent equilibrium calculations, resulting in speedups of almost two orders of magnitude. We remark that our new on-demand machine learning method can be applied to any case in which a massive number of sequential/parallel evaluations of a computationally expensive function $f$ needs to be done, $y=f(x)$. We remark, that, in contrast to traditional machine learning algorithms, our on-demand training approach does not require a statistics-based training phase before the actual simulation of interest commences. The introduced on-demand training scheme requires, however, the first-order derivatives $\partial f/\partial x$ for later smart predictions.