 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnsemble-Based Graph Representation of fMRI Data for Cognitive Brain State Classification
Aug 08, 2025Understanding and classifying human cognitive brain states based on neuroimaging data remains one of the foremost and most challenging problems in neuroscience, owing to the high dimensionality and intrinsic noise of the signals. In this work, we propose an ensemble-based graph representation method of functional magnetic resonance imaging (fMRI) data for the task of binary brain-state classification. Our method builds the graph by leveraging multiple base machine-learning models: each edge weight reflects the difference in posterior probabilities between two cognitive states, yielding values in the range [-1, 1] that encode confidence in a given state. We applied this approach to seven cognitive tasks from the Human Connectome Project (HCP 1200 Subject Release), including working memory, gambling, motor activity, language, social cognition, relational processing, and emotion processing. Using only the mean incident edge weights of the graphs as features, a simple logistic-regression classifier achieved average accuracies from 97.07% to 99.74%. We also compared our ensemble graphs with classical correlation-based graphs in a classification task with a graph neural network (GNN). In all experiments, the highest classification accuracy was obtained with ensemble graphs. These results demonstrate that ensemble graphs convey richer topological information and enhance brain-state discrimination. Our approach preserves edge-level interpretability of the fMRI graph representation, is adaptable to multiclass and regression tasks, and can be extended to other neuroimaging modalities and pathological-state classification.
Astrocytes mediate analogous memory in a multi-layer neuron-astrocytic network
Aug 31, 2021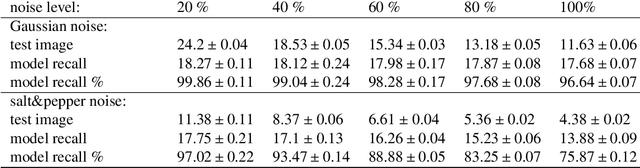



Modeling the neuronal processes underlying short-term working memory remains the focus of many theoretical studies in neuroscience. Here we propose a mathematical model of spiking neuron network (SNN) demonstrating how a piece of information can be maintained as a robust activity pattern for several seconds then completely disappear if no other stimuli come. Such short-term memory traces are preserved due to the activation of astrocytes accompanying the SNN. The astrocytes exhibit calcium transients at a time scale of seconds. These transients further modulate the efficiency of synaptic transmission and, hence, the firing rate of neighboring neurons at diverse timescales through gliotransmitter release. We show how such transients continuously encode frequencies of neuronal discharges and provide robust short-term storage of analogous information. This kind of short-term memory can keep operative information for seconds, then completely forget it to avoid overlapping with forthcoming patterns. The SNN is inter-connected with the astrocytic layer by local inter-cellular diffusive connections. The astrocytes are activated only when the neighboring neurons fire quite synchronously, e.g. when an information pattern is loaded. For illustration, we took greyscale photos of people's faces where the grey level encoded the level of applied current stimulating the neurons. The astrocyte feedback modulates (facilitates) synaptic transmission by varying the frequency of neuronal firing. We show how arbitrary patterns can be loaded, then stored for a certain interval of time, and retrieved if the appropriate clue pattern is applied to the input.
Detection of Epigenomic Network Community Oncomarkers
Aug 01, 2016

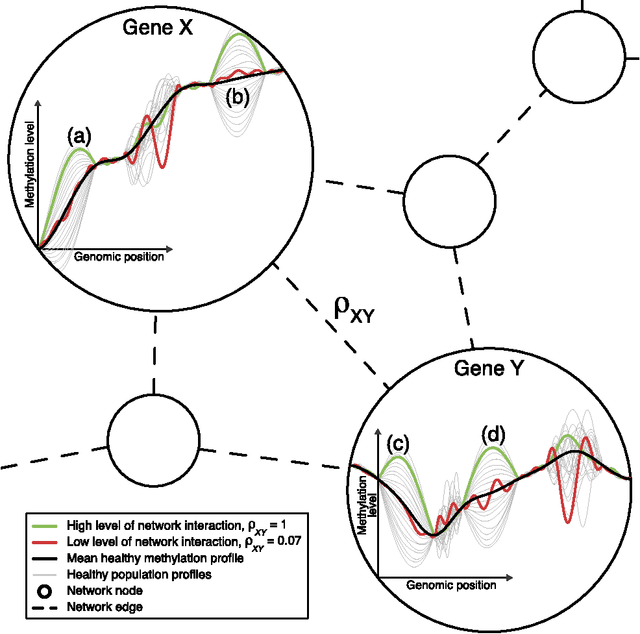

In this paper we propose network methodology to infer prognostic cancer biomarkers based on the epigenetic pattern DNA methylation. Epigenetic processes such as DNA methylation reflect environmental risk factors, and are increasingly recognised for their fundamental role in diseases such as cancer. DNA methylation is a gene-regulatory pattern, and hence provides a means by which to assess genomic regulatory interactions. Network models are a natural way to represent and analyse groups of such interactions. The utility of network models also increases as the quantity of data and number of variables increase, making them increasingly relevant to large-scale genomic studies. We propose methodology to infer prognostic genomic networks from a DNA methylation-based measure of genomic interaction and association. We then show how to identify prognostic biomarkers from such networks, which we term `network community oncomarkers'. We illustrate the power of our proposed methodology in the context of a large publicly available breast cancer dataset.