 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTransferring Chemical and Energetic Knowledge Between Molecular Systems with Machine Learning
Paper and Code
May 06, 2022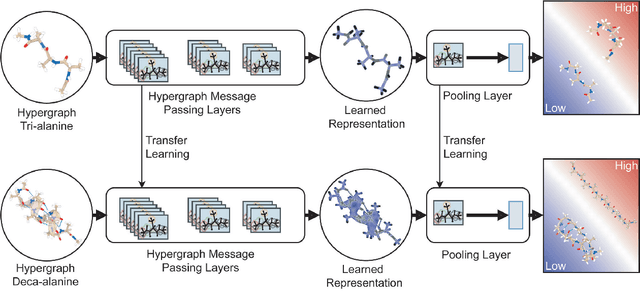
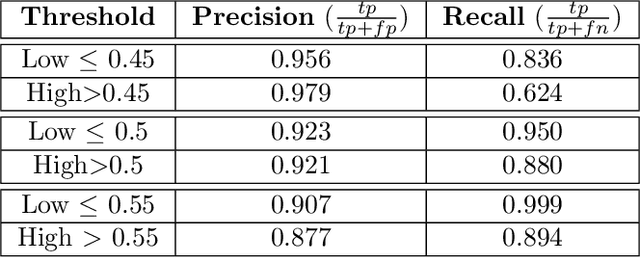
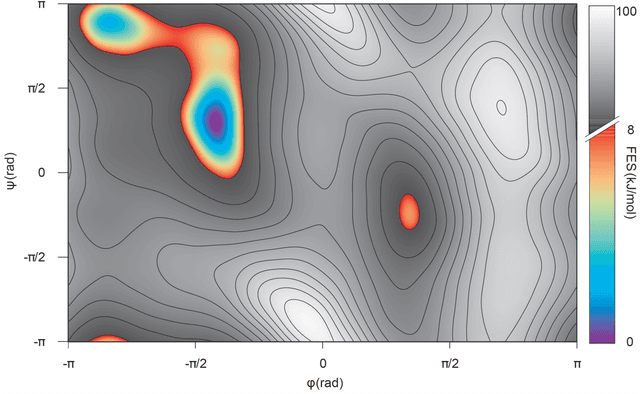
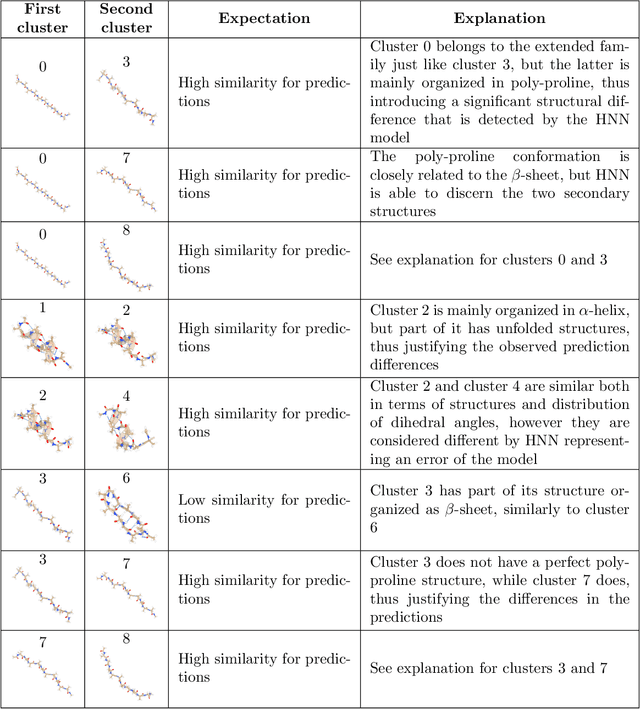
Predicting structural and energetic properties of a molecular system is one of the fundamental tasks in molecular simulations, and it has use cases in chemistry, biology, and medicine. In the past decade, the advent of machine learning algorithms has impacted on molecular simulations for various tasks, including property prediction of atomistic systems. In this paper, we propose a novel methodology for transferring knowledge obtained from simple molecular systems to a more complex one, possessing a significantly larger number of atoms and degrees of freedom. In particular, we focus on the classification of high and low free-energy states. Our approach relies on utilizing (i) a novel hypergraph representation of molecules, encoding all relevant information for characterizing the potential energy of a conformation, and (ii) novel message passing and pooling layers for processing and making predictions on such hypergraph-structured data. Despite the complexity of the problem, our results show a remarkable AUC of 0.92 for transfer learning from tri-alanine to the deca-alanine system. Moreover, we show that the very same transfer learning approach can be used to group, in an unsupervised way, various secondary structures of deca-alanine in clusters having similar free-energy values. Our study represents a proof of concept that reliable transfer learning models for molecular systems can be designed paving the way to unexplored routes in prediction of structural and energetic properties of biologically relevant systems.