 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards fully automated protein structure elucidation with NMR spectroscopy
Paper and Code
Jul 31, 2018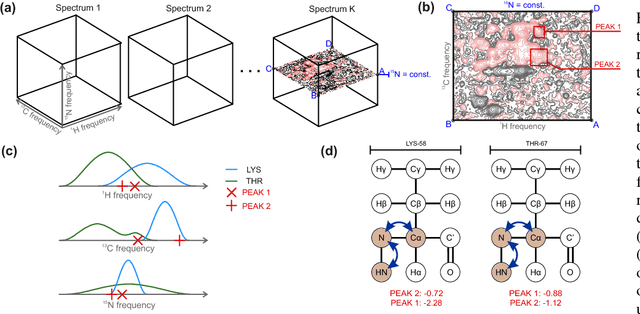
Nuclear magnetic resonance (NMR) spectroscopy is one of the leading techniques for protein studies. The method features a number of properties, allowing to explain macromolecular interactions mechanistically and resolve structures with atomic resolution. However, due to laborious data analysis, a full potential of NMR spectroscopy remains unexploited. Here we present an approach aiming at automation of two major bottlenecks in the analysis pipeline, namely, peak picking and chemical shift assignment. Our approach combines deep learning, non-parametric models and combinatorial optimization, and is able to detect signals of interest in a multidimensional NMR data with high accuracy and match them with atoms in medium-length protein sequences, which is a preliminary step to solve protein spatial structure.