 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards Evolutionary-based Automated Machine Learning for Small Molecule Pharmacokinetic Prediction
Paper and Code
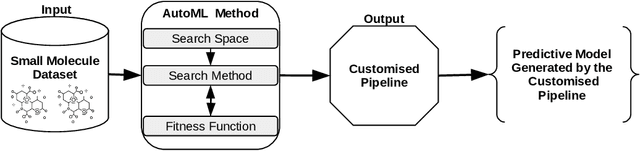
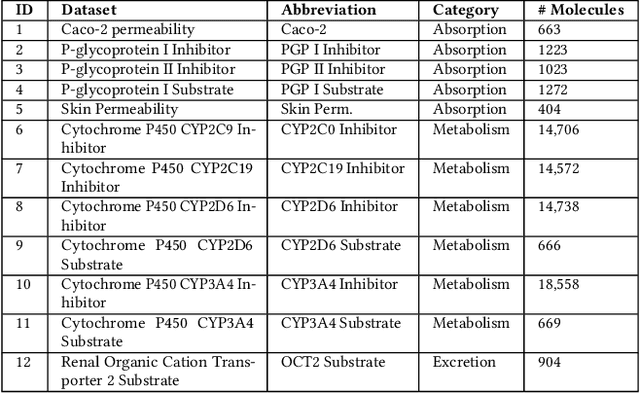
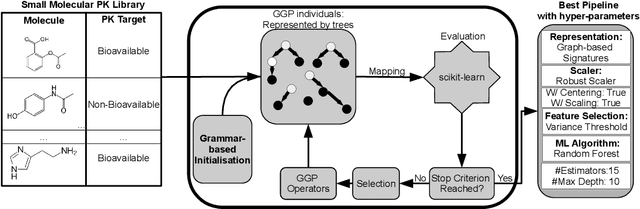
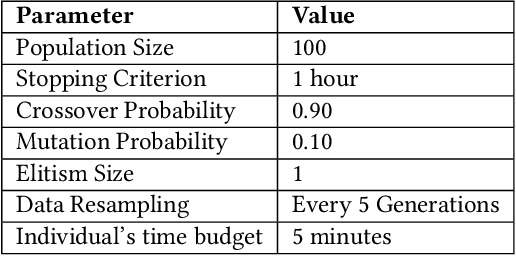
Machine learning (ML) is revolutionising drug discovery by expediting the prediction of small molecule properties essential for developing new drugs. These properties -- including absorption, distribution, metabolism and excretion (ADME)-- are crucial in the early stages of drug development since they provide an understanding of the course of the drug in the organism, i.e., the drug's pharmacokinetics. However, existing methods lack personalisation and rely on manually crafted ML algorithms or pipelines, which can introduce inefficiencies and biases into the process. To address these challenges, we propose a novel evolutionary-based automated ML method (AutoML) specifically designed for predicting small molecule properties, with a particular focus on pharmacokinetics. Leveraging the advantages of grammar-based genetic programming, our AutoML method streamlines the process by automatically selecting algorithms and designing predictive pipelines tailored to the particular characteristics of input molecular data. Results demonstrate AutoML's effectiveness in selecting diverse ML algorithms, resulting in comparable or even improved predictive performances compared to conventional approaches. By offering personalised ML-driven pipelines, our method promises to enhance small molecule research in drug discovery, providing researchers with a valuable tool for accelerating the development of novel therapeutic drugs.