 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable neural quantum states architecture for quantum chemistry
Paper and Code
Aug 11, 2022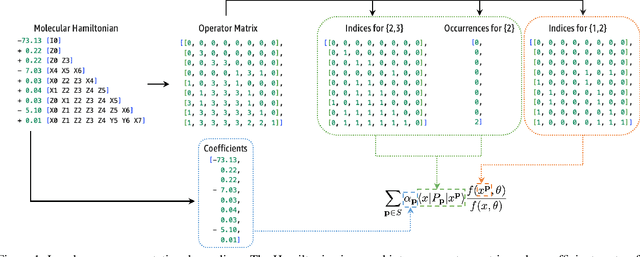
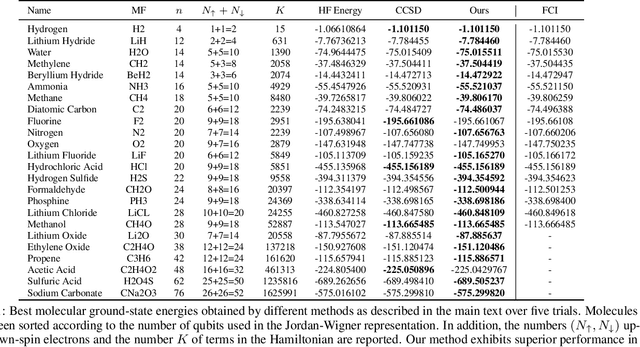
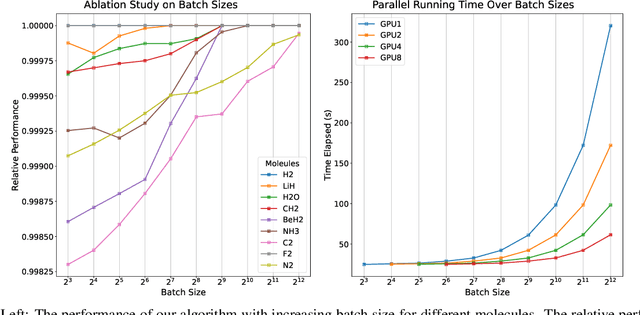
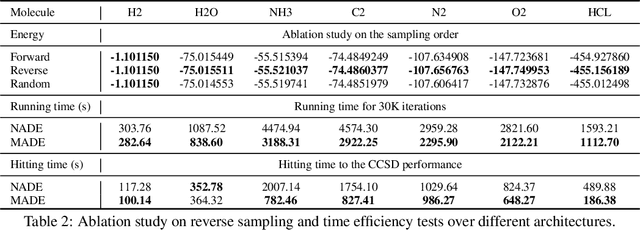
Variational optimization of neural-network representations of quantum states has been successfully applied to solve interacting fermionic problems. Despite rapid developments, significant scalability challenges arise when considering molecules of large scale, which correspond to non-locally interacting quantum spin Hamiltonians consisting of sums of thousands or even millions of Pauli operators. In this work, we introduce scalable parallelization strategies to improve neural-network-based variational quantum Monte Carlo calculations for ab-initio quantum chemistry applications. We establish GPU-supported local energy parallelism to compute the optimization objective for Hamiltonians of potentially complex molecules. Using autoregressive sampling techniques, we demonstrate systematic improvement in wall-clock timings required to achieve CCSD baseline target energies. The performance is further enhanced by accommodating the structure of resultant spin Hamiltonians into the autoregressive sampling ordering. The algorithm achieves promising performance in comparison with the classical approximate methods and exhibits both running time and scalability advantages over existing neural-network based methods.