 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScaffold Embeddings: Learning the Structure Spanned by Chemical Fragments, Scaffolds and Compounds
Paper and Code
Mar 11, 2021


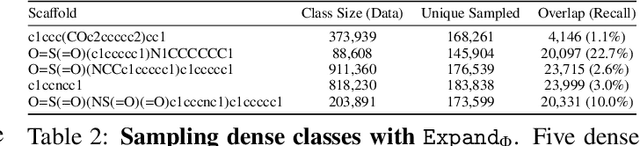
Molecules have seemed like a natural fit to deep learning's tendency to handle a complex structure through representation learning, given enough data. However, this often continuous representation is not natural for understanding chemical space as a domain and is particular to samples and their differences. We focus on exploring a natural structure for representing chemical space as a structured domain: embedding drug-like chemical space into an enumerable hypergraph based on scaffold classes linked through an inclusion operator. This paper shows how molecules form classes of scaffolds, how scaffolds relate to each in a hypergraph, and how this structure of scaffolds is natural for drug discovery workflows such as predicting properties and optimizing molecular structures. We compare the assumptions and utility of various embeddings of molecules, such as their respective induced distance metrics, their extendibility to represent chemical space as a structured domain, and the consequences of utilizing the structure for learning tasks.