 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQuantitative Prediction on the Enantioselectivity of Multiple Chiral Iodoarene Scaffolds Based on Whole Geometry
Paper and Code
Mar 25, 2021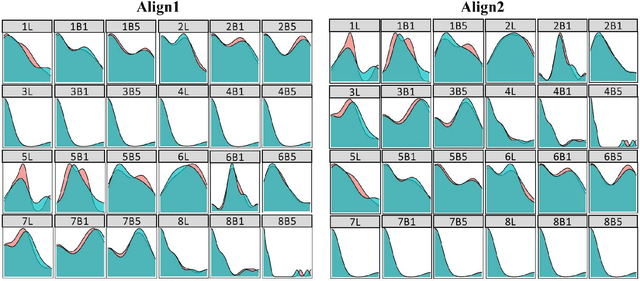
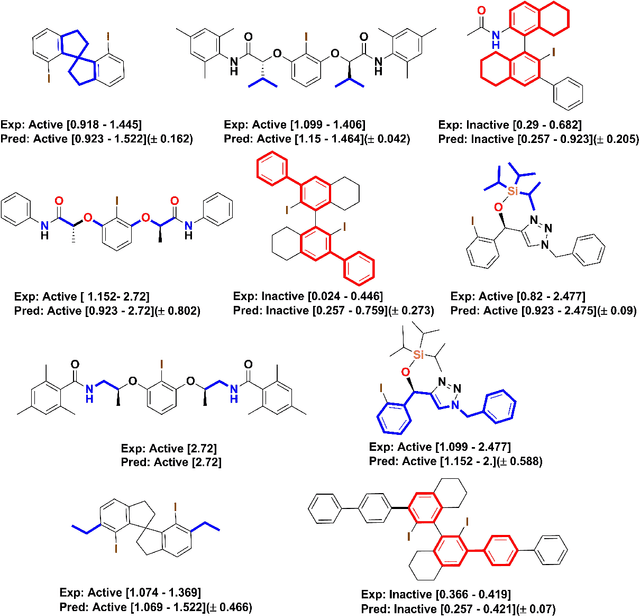
The mechanistic underpinnings of asymmetric catalysis at atomic levels provide shortcuts for developing the potential value of chiral catalysts beyond the current state-of-the-art. In the enantioselective redox transformations, the present intuition-driven studies require a systematic approach to support their intuitive idea. Arguably, the most systematic approach would be based on the reliable quantitative structure-selectivity relationship of diverse and dissimilar chiral scaffolds in an optimal feature space that is universally applied to reactions. Here, we introduce a predictive workflow for the extension of the reaction scope of chiral catalysts across name reactions. For this purpose, whole geometry descriptors were encoded from DFT optimized 3D structures of multiple catalyst scaffolds, 113 catalysts in 9 clusters. The molecular descriptors were verified by the statistical comparison of the enantioselective predictive classification models built from each descriptors of chiral iodoarenes. More notably, capturing the whole molecular geometry through one hot encoding of split three-dimensional molecular fingerprints presented reliable enantioselective predictive regression models for three different name reactions by recycling the data and metadata obtained across reactions. The potential use value of this workflow and the advantages of recyclability, compatibility, and generality proved that the workflow can be applied for name reactions other than the aforementioned name reactions (out of samples). Furthermore, for the consensus prediction of ensemble models, this global descriptor can be compared with sterimol parameters and noncovalent interaction vectors. This study is one case showing how to overcome the sparsity of experimental data in organic reactions, especially asymmetric catalysis.