 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting drug properties with parameter-free machine learning: Pareto-Optimal Embedded Modeling (POEM)
Paper and Code
Feb 11, 2020
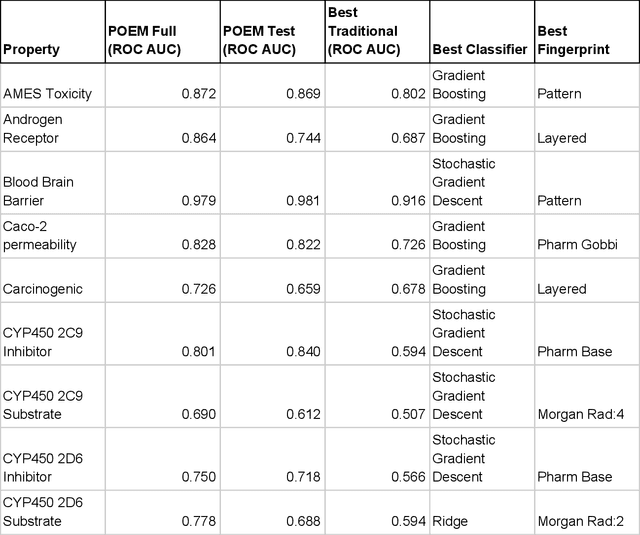


The prediction of absorption, distribution, metabolism, excretion, and toxicity (ADMET) of small molecules from their molecular structure is a central problem in medicinal chemistry with great practical importance in drug discovery. Creating predictive models conventionally requires substantial trial-and-error for the selection of molecular representations, machine learning (ML) algorithms, and hyperparameter tuning. A generally applicable method that performs well on all datasets without tuning would be of great value but is currently lacking. Here, we describe Pareto-Optimal Embedded Modeling (POEM), a similarity-based method for predicting molecular properties. POEM is a non-parametric, supervised ML algorithm developed to generate reliable predictive models without need for optimization. POEMs predictive strength is obtained by combining multiple different representations of molecular structures in a context-specific manner, while maintaining low dimensionality. We benchmark POEM relative to industry-standard ML algorithms and published results across 17 classifications tasks. POEM performs well in all cases and reduces the risk of overfitting.