 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePan-cancer gene set discovery via scRNA-seq for optimal deep learning based downstream tasks
Paper and Code
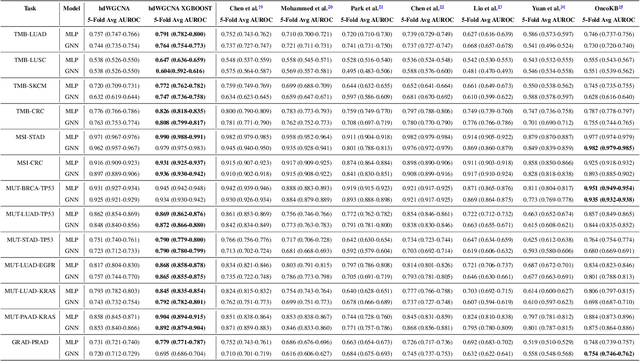
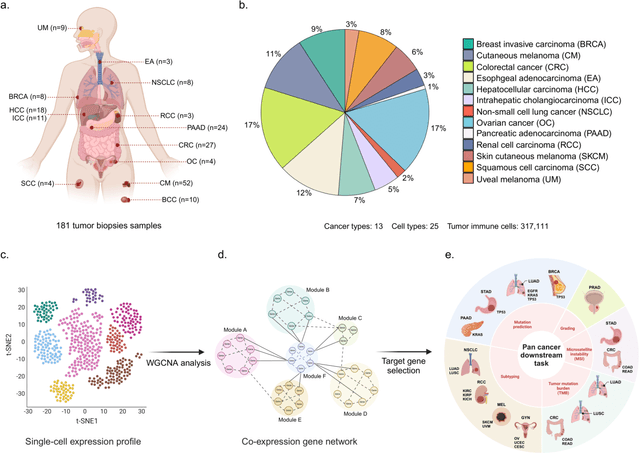
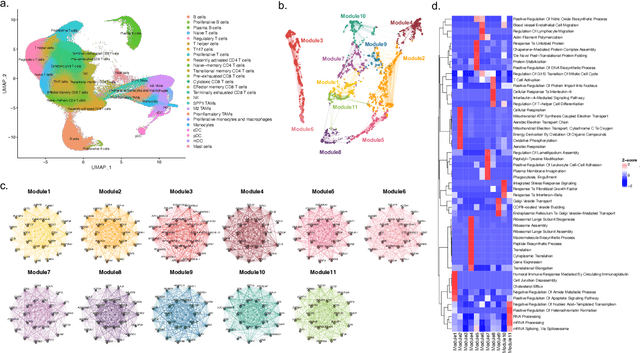
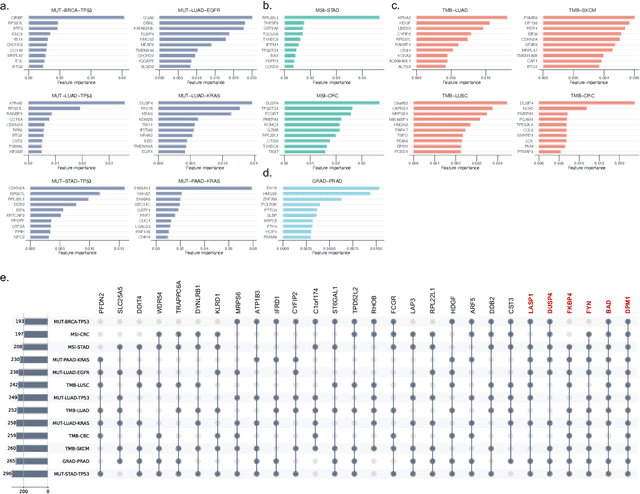
The application of machine learning to transcriptomics data has led to significant advances in cancer research. However, the high dimensionality and complexity of RNA sequencing (RNA-seq) data pose significant challenges in pan-cancer studies. This study hypothesizes that gene sets derived from single-cell RNA sequencing (scRNA-seq) data will outperform those selected using bulk RNA-seq in pan-cancer downstream tasks. We analyzed scRNA-seq data from 181 tumor biopsies across 13 cancer types. High-dimensional weighted gene co-expression network analysis (hdWGCNA) was performed to identify relevant gene sets, which were further refined using XGBoost for feature selection. These gene sets were applied to downstream tasks using TCGA pan-cancer RNA-seq data and compared to six reference gene sets and oncogenes from OncoKB evaluated with deep learning models, including multilayer perceptrons (MLPs) and graph neural networks (GNNs). The XGBoost-refined hdWGCNA gene set demonstrated higher performance in most tasks, including tumor mutation burden assessment, microsatellite instability classification, mutation prediction, cancer subtyping, and grading. In particular, genes such as DPM1, BAD, and FKBP4 emerged as important pan-cancer biomarkers, with DPM1 consistently significant across tasks. This study presents a robust approach for feature selection in cancer genomics by integrating scRNA-seq data and advanced analysis techniques, offering a promising avenue for improving predictive accuracy in cancer research.