 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMoleHD: Automated Drug Discovery using Brain-Inspired Hyperdimensional Computing
Paper and Code
Jun 05, 2021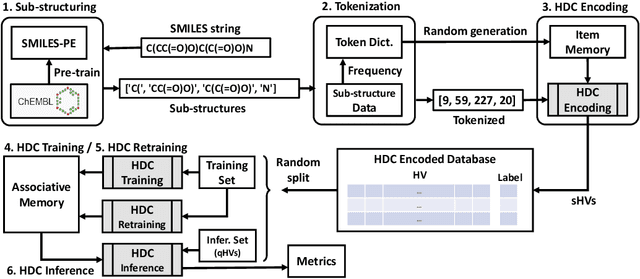
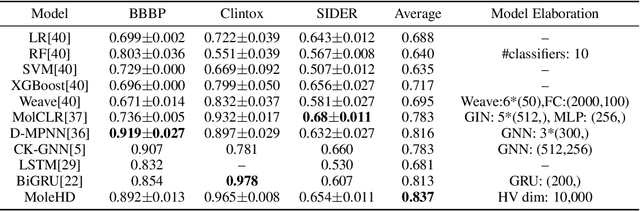
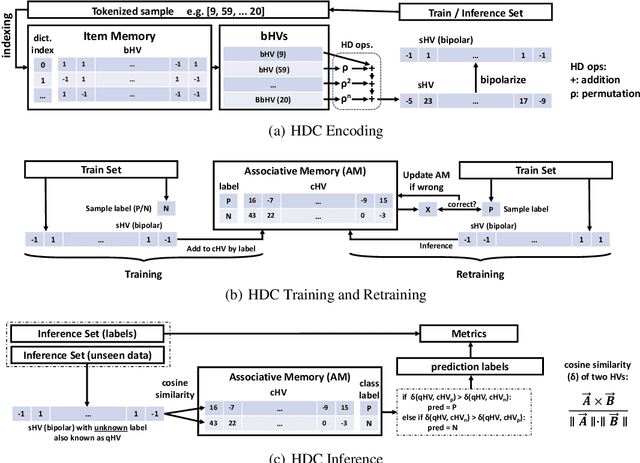
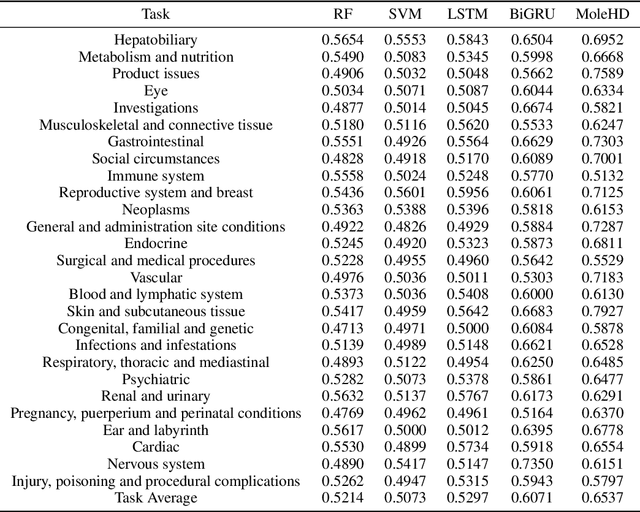
Modern drug discovery is often time-consuming, complex and cost-ineffective due to the large volume of molecular data and complicated molecular properties. Recently, machine learning algorithms have shown promising results in virtual screening of automated drug discovery by predicting molecular properties. While emerging learning methods such as graph neural networks and recurrent neural networks exhibit high accuracy, they are also notoriously computation-intensive and memory-intensive with operations such as feature embeddings or deep convolutions. In this paper, we propose a viable alternative to neural network classifiers. We present MoleHD, a method based on brain-inspired hyperdimensional computing (HDC) for molecular property prediction. We first transform the SMILES presentation of molecules into feature vectors by SMILE-PE tokenizers pretrained on the ChEMBL database. Then, we develop HDC encoders to project such features into high-dimensional vectors that are used for training and inference. We perform an extensive evaluation using 30 classification tasks from 3 widely-used molecule datasets and compare MoleHD with 10 baseline methods including 6 SOTA neural network classifiers. Results show that MoleHD is able to outperform all the baseline methods on average across 30 classification tasks with significantly reduced computing cost. To the best of our knowledge, we develop the first HDC-based method for drug discovery. The promising results presented in this paper can potentially lead to a novel path in drug discovery research.