 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeModelAngelo: Automated Model Building in Cryo-EM Maps
Paper and Code
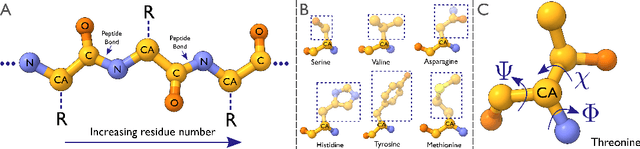
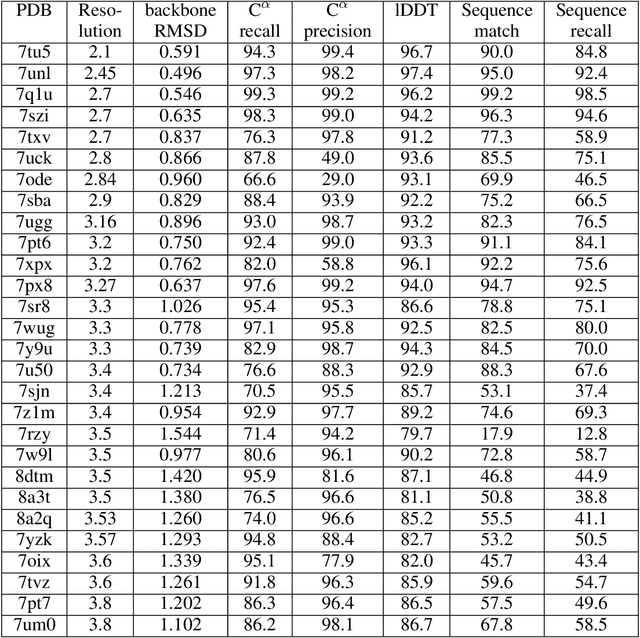
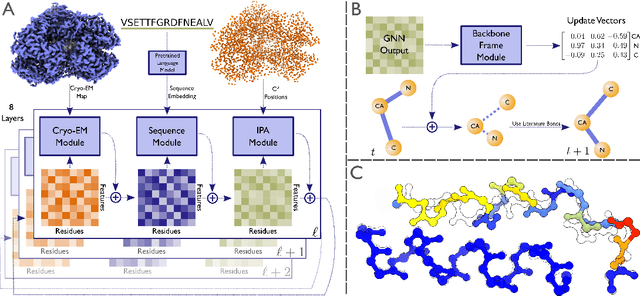
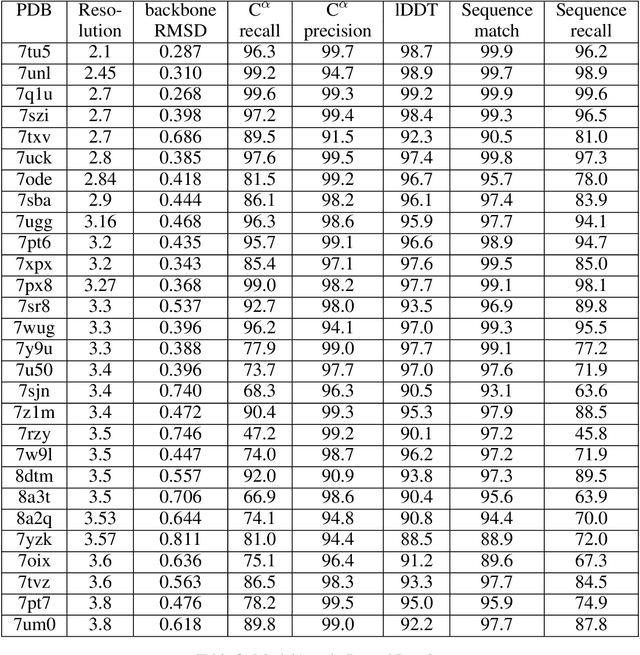
Electron cryo-microscopy (cryo-EM) produces three-dimensional (3D) maps of the electrostatic potential of biological macromolecules, including proteins. At sufficient resolution, the cryo-EM maps, along with some knowledge about the imaged molecules, allow de novo atomic modelling. Typically, this is done through a laborious manual process. Recent advances in machine learning applications to protein structure prediction show potential for automating this process. Taking inspiration from these techniques, we have built ModelAngelo for automated model building of proteins in cryo-EM maps. ModelAngelo first uses a residual convolutional neural network (CNN) to initialize a graph representation with nodes assigned to individual amino acids of the proteins in the map and edges representing the protein chain. The graph is then refined with a graph neural network (GNN) that combines the cryo-EM data, the amino acid sequence data and prior knowledge about protein geometries. The GNN refines the geometry of the protein chain and classifies the amino acids for each of its nodes. The final graph is post-processed with a hidden Markov model (HMM) search to map each protein chain to entries in a user provided sequence file. Application to 28 test cases shows that ModelAngelo outperforms the state-of-the-art and approximates manual building for cryo-EM maps with resolutions better than 3.5 \r{A}.