 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLow-cost machine learning approach to the prediction of transition metal phosphor excited state properties
Paper and Code
Sep 18, 2022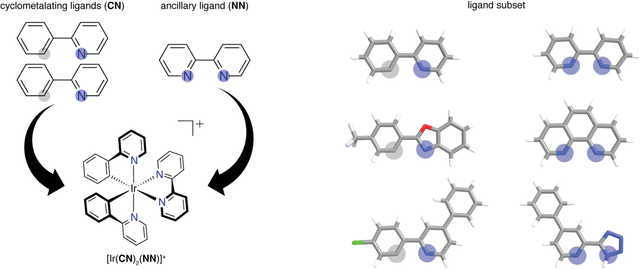
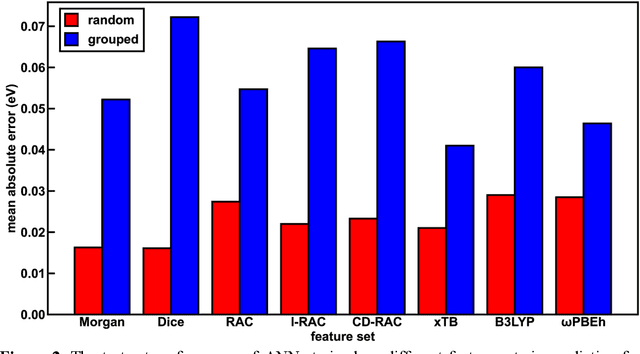
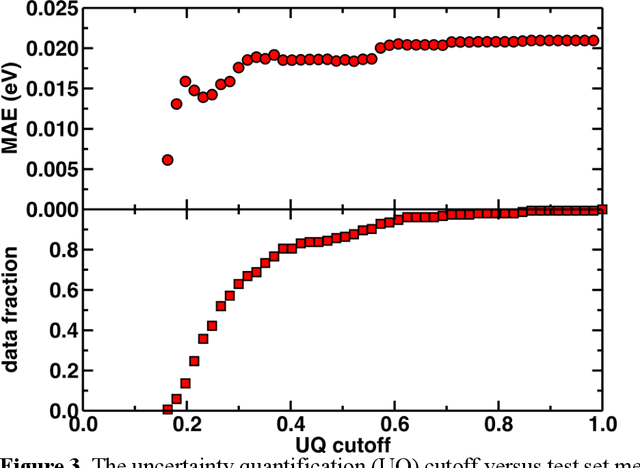
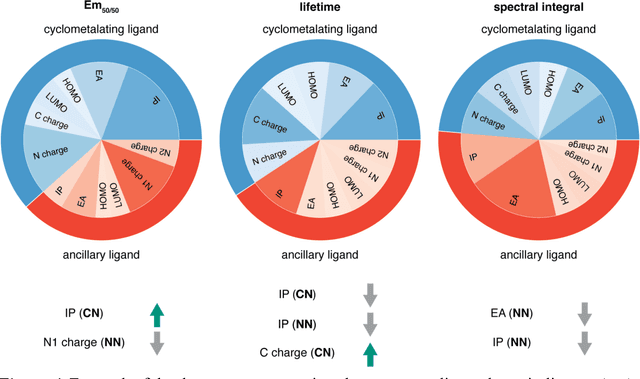
Photoactive iridium complexes are of broad interest due to their applications ranging from lighting to photocatalysis. However, the excited state property prediction of these complexes challenges ab initio methods such as time-dependent density functional theory (TDDFT) both from an accuracy and a computational cost perspective, complicating high throughput virtual screening (HTVS). We instead leverage low-cost machine learning (ML) models to predict the excited state properties of photoactive iridium complexes. We use experimental data of 1,380 iridium complexes to train and evaluate the ML models and identify the best-performing and most transferable models to be those trained on electronic structure features from low-cost density functional theory tight binding calculations. Using these models, we predict the three excited state properties considered, mean emission energy of phosphorescence, excited state lifetime, and emission spectral integral, with accuracy competitive with or superseding TDDFT. We conduct feature importance analysis to identify which iridium complex attributes govern excited state properties and we validate these trends with explicit examples. As a demonstration of how our ML models can be used for HTVS and the acceleration of chemical discovery, we curate a set of novel hypothetical iridium complexes and identify promising ligands for the design of new phosphors.