 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInjecting Domain Knowledge from Empirical Interatomic Potentials to Neural Networks for Predicting Material Properties
Paper and Code
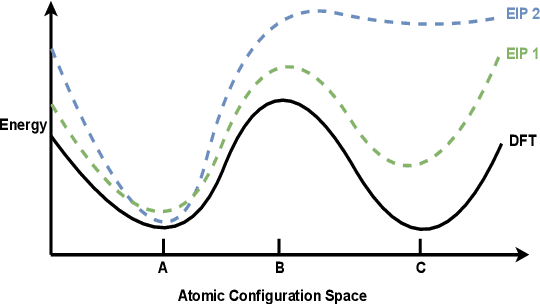
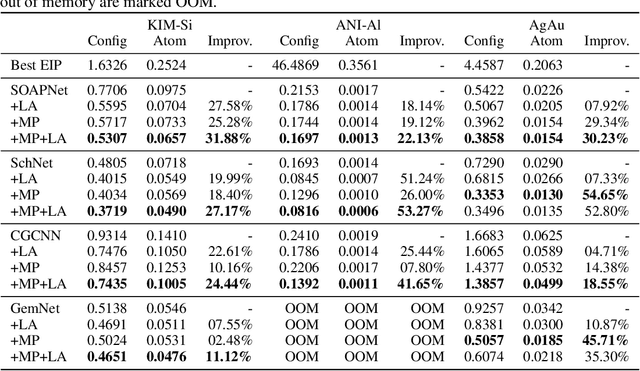
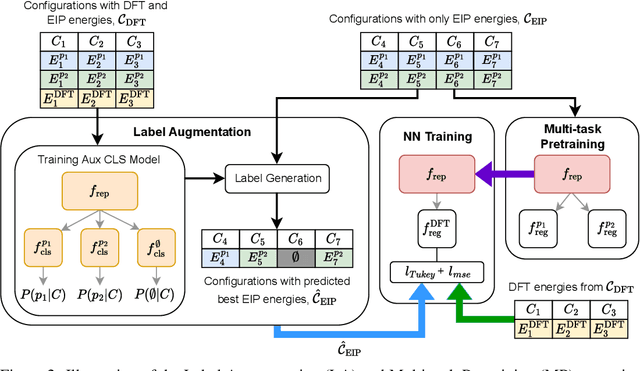
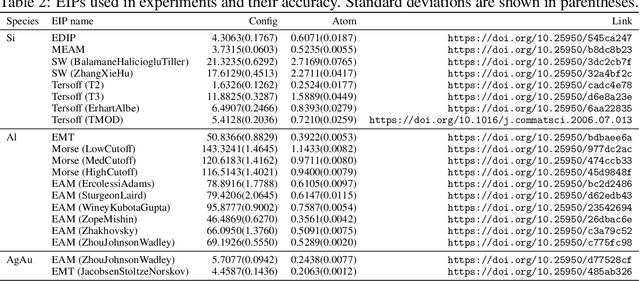
For decades, atomistic modeling has played a crucial role in predicting the behavior of materials in numerous fields ranging from nanotechnology to drug discovery. The most accurate methods in this domain are rooted in first-principles quantum mechanical calculations such as density functional theory (DFT). Because these methods have remained computationally prohibitive, practitioners have traditionally focused on defining physically motivated closed-form expressions known as empirical interatomic potentials (EIPs) that approximately model the interactions between atoms in materials. In recent years, neural network (NN)-based potentials trained on quantum mechanical (DFT-labeled) data have emerged as a more accurate alternative to conventional EIPs. However, the generalizability of these models relies heavily on the amount of labeled training data, which is often still insufficient to generate models suitable for general-purpose applications. In this paper, we propose two generic strategies that take advantage of unlabeled training instances to inject domain knowledge from conventional EIPs to NNs in order to increase their generalizability. The first strategy, based on weakly supervised learning, trains an auxiliary classifier on EIPs and selects the best-performing EIP to generate energies to supplement the ground-truth DFT energies in training the NN. The second strategy, based on transfer learning, first pretrains the NN on a large set of easily obtainable EIP energies, and then fine-tunes it on ground-truth DFT energies. Experimental results on three benchmark datasets demonstrate that the first strategy improves baseline NN performance by 5% to 51% while the second improves baseline performance by up to 55%. Combining them further boosts performance.