 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHybrid Quantum Generative Adversarial Networks for Molecular Simulation and Drug Discovery
Paper and Code
Dec 15, 2022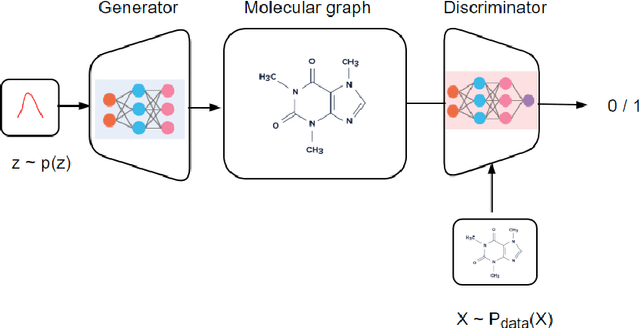

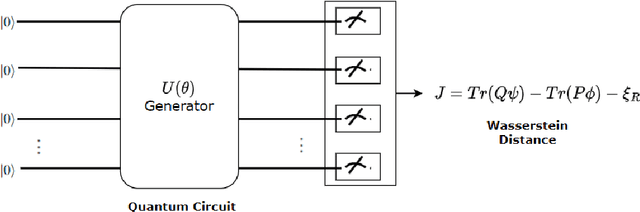

In molecular research, simulation \& design of molecules are key areas with significant implications for drug development, material science, and other fields. Current classical computational power falls inadequate to simulate any more than small molecules, let alone protein chains on hundreds of peptide. Therefore these experiment are done physically in wet-lab, but it takes a lot of time \& not possible to examine every molecule due to the size of the search area, tens of billions of dollars are spent every year in these research experiments. Molecule simulation \& design has lately advanced significantly by machine learning models, A fresh perspective on the issue of chemical synthesis is provided by deep generative models for graph-structured data. By optimising differentiable models that produce molecular graphs directly, it is feasible to avoid costly search techniques in the discrete and huge space of chemical structures. But these models also suffer from computational limitations when dimensions become huge and consume huge amount of resources. Quantum Generative machine learning in recent years have shown some empirical results promising significant advantages over classical counterparts.