 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGene Shaving using influence function of a kernel method
Paper and Code
Sep 05, 2018
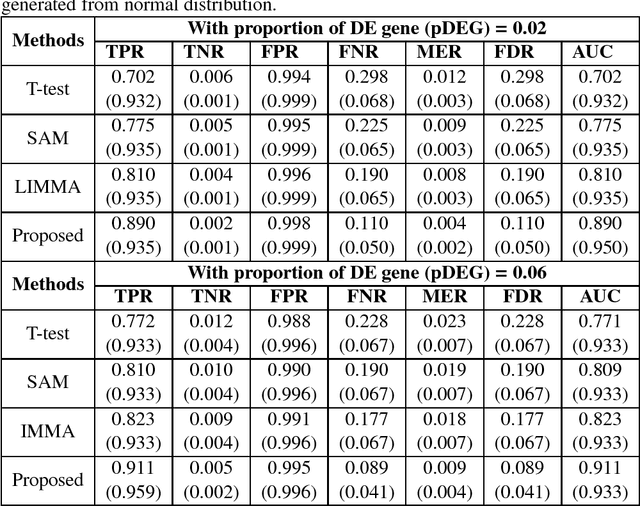

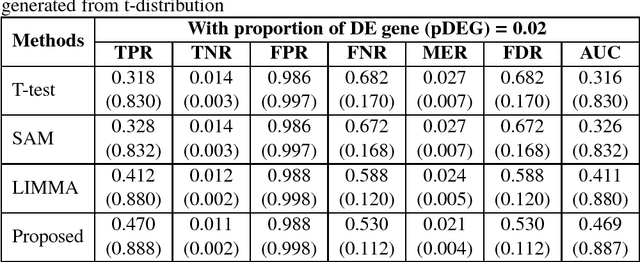
Identifying significant subsets of the genes, gene shaving is an essential and challenging issue for biomedical research for a huge number of genes and the complex nature of biological networks,. Since positive definite kernel based methods on genomic information can improve the prediction of diseases, in this paper we proposed a new method, "kernel gene shaving (kernel canonical correlation analysis (kernel CCA) based gene shaving). This problem is addressed using the influence function of the kernel CCA. To investigate the performance of the proposed method in a comparison of three popular gene selection methods (T-test, SAM and LIMMA), we were used extensive simulated and real microarray gene expression datasets. The performance measures AUC was computed for each of the methods. The achievement of the proposed method has improved than the three well-known gene selection methods. In real data analysis, the proposed method identified a subsets of $210$ genes out of $2000$ genes. The network of these genes has significantly more interactions than expected, which indicates that they may function in a concerted effort on colon cancer.