 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEncoding protein dynamic information in graph representation for functional residue identification
Paper and Code
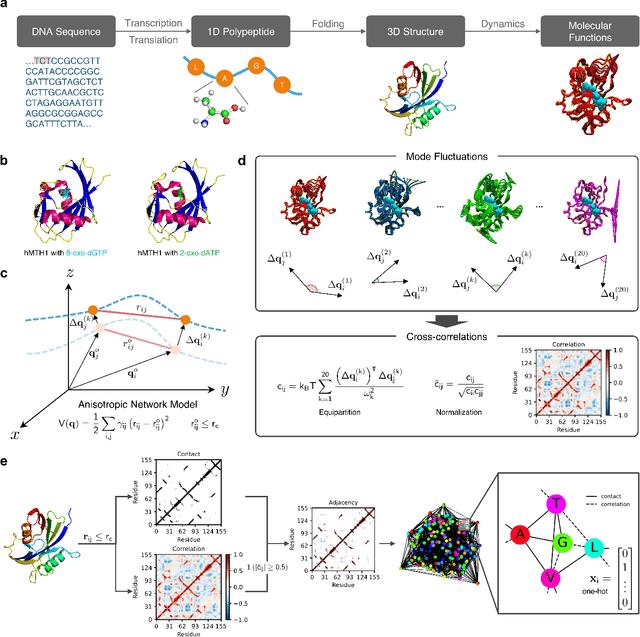



Recent advances in protein function prediction exploit graph-based deep learning approaches to correlate the structural and topological features of proteins with their molecular functions. However, proteins in vivo are not static but dynamic molecules that alter conformation for functional purposes. Here we apply normal mode analysis to native protein conformations and augment protein graphs by connecting edges between dynamically correlated residue pairs. In the multilabel function classification task, our method demonstrates a remarkable performance gain based on this dynamics-informed representation. The proposed graph neural network, ProDAR, increases the interpretability and generalizability of residue-level annotations and robustly reflects structural nuance in proteins. We elucidate the importance of dynamic information in graph representation by comparing class activation maps for the hMTH1, nitrophorin, and SARS-CoV-2 receptor binding domain. Our model successfully learns the dynamic fingerprints of proteins and provides molecular insights into protein functions, with vast untapped potential for broad biotechnology and pharmaceutical applications.