 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeep learning is competing random forest in computational docking
Paper and Code
Aug 23, 2016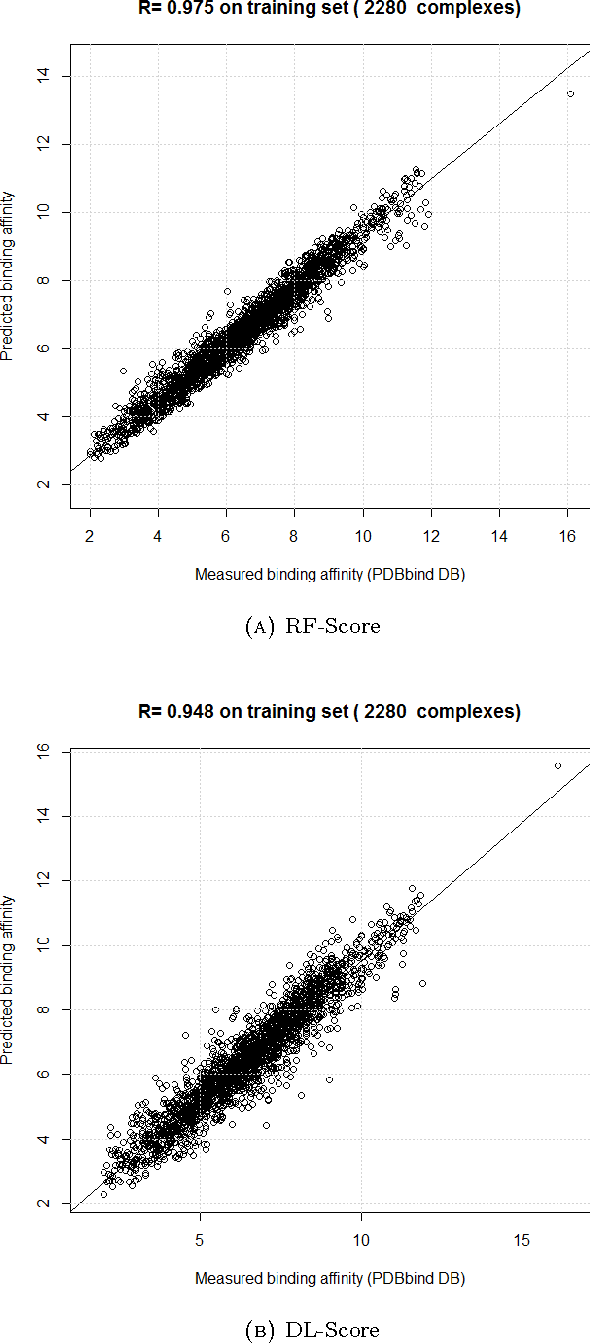
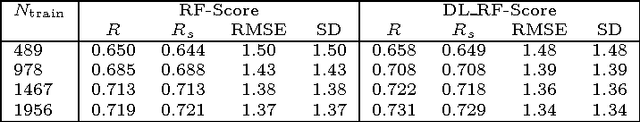
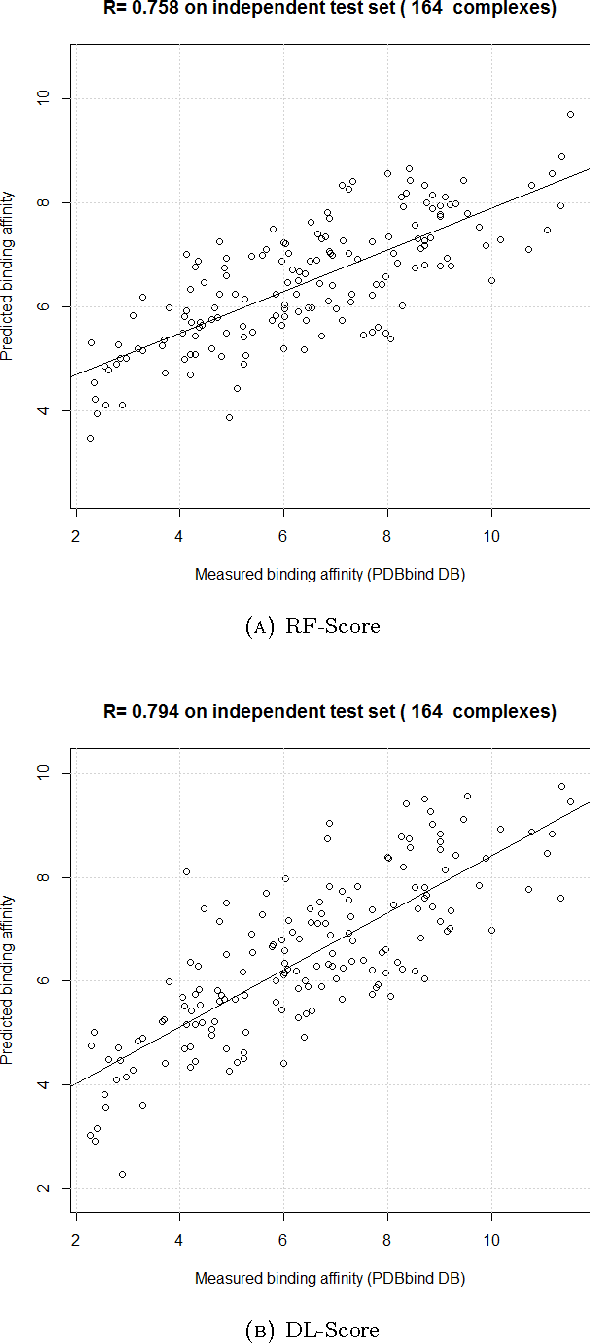
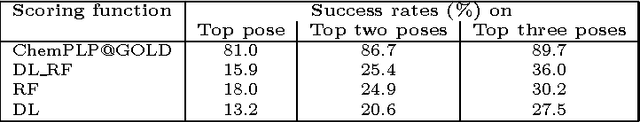
Computational docking is the core process of computer-aided drug design; it aims at predicting the best orientation and conformation of a small drug molecule when bound to a target large protein receptor. The docking quality is typically measured by a scoring function: a mathematical predictive model that produces a score representing the binding free energy and hence the stability of the resulting complex molecule. We analyze the performance of both learning techniques on the scoring power, the ranking power, docking power, and screening power using the PDBbind 2013 database. For the scoring and ranking powers, the proposed learning scoring functions depend on a wide range of features (energy terms, pharmacophore, intermolecular) that entirely characterize the protein-ligand complexes. For the docking and screening powers, the proposed learning scoring functions depend on the intermolecular features of the RF-Score to utilize a larger number of training complexes. For the scoring power, the DL\_RF scoring function achieves Pearson's correlation coefficient between the predicted and experimentally measured binding affinities of 0.799 versus 0.758 of the RF scoring function. For the ranking power, the DL scoring function ranks the ligands bound to fixed target protein with accuracy 54% for the high-level ranking and with accuracy 78% for the low-level ranking while the RF scoring function achieves (46% and 62%) respectively. For the docking power, the DL\_RF scoring function has a success rate when the three best-scored ligand binding poses are considered within 2 \AA\ root-mean-square-deviation from the native pose of 36.0% versus 30.2% of the RF scoring function. For the screening power, the DL scoring function has an average enrichment factor and success rate at the top 1% level of (2.69 and 6.45%) respectively versus (1.61 and 4.84%) respectively of the RF scoring function.