 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChemistry-informed Macromolecule Graph Representation for Similarity Computation and Supervised Learning
Paper and Code
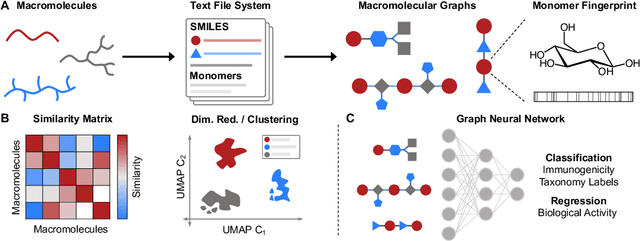
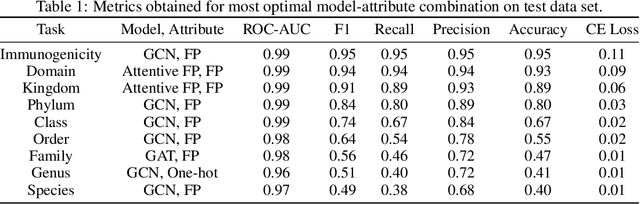
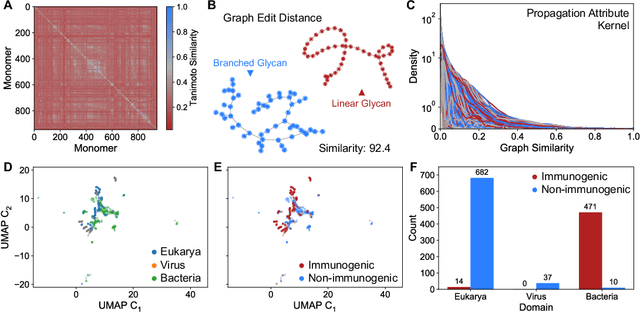
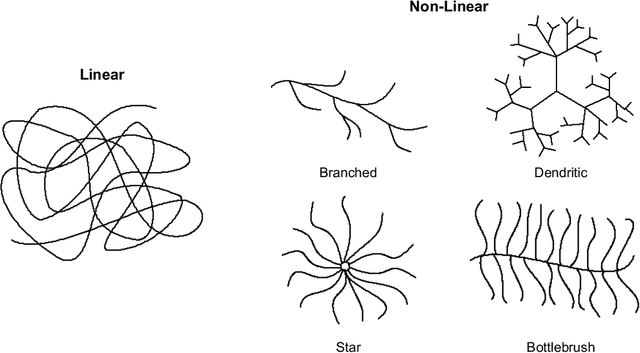
Macromolecules are large, complex molecules composed of covalently bonded monomer units, existing in different stereochemical configurations and topologies. As a result of such chemical diversity, representing, comparing, and learning over macromolecules emerge as critical challenges. To address this, we developed a macromolecule graph representation, with monomers and bonds as nodes and edges, respectively. We captured the inherent chemistry of the macromolecule by using molecular fingerprints for node and edge attributes. For the first time, we demonstrated computation of chemical similarity between 2 macromolecules of varying chemistry and topology, using exact graph edit distances and graph kernels. We also trained graph neural networks for a variety of glycan classification tasks, achieving state-of-the-art results. Our work has two-fold implications - it provides a general framework for representation, comparison, and learning of macromolecules; and enables quantitative chemistry-informed decision-making and iterative design in the macromolecular chemical space.